The Genetic Basis of Cancer
J. Michael Bishop
 J. Michael Bishop
J. Michael Bishop
J. Michael Bishop is University Professor Emeritus and Director Emeritus of the G.W. Hooper Research Foundation at the University of California, San Francisco (UCSF). He is a graduate of Gettysburg College and Harvard Medical School. After postdoctoral training at the National Institutes of Health and a year as a visiting scientist in Hamburg, Germany, he joined the faculty of UCSF in 1968. He has devoted his career to research on the molecular pathogenesis of cancer, the teaching of medical and graduate students, and service as chancellor of UCSF from 1998 to 2009. Dr. Bishop’s honors include the Albert Lasker Basic Medical Research Award, the U.S. National Medal of Science, the Nobel Prize for Physiology or Medicine, multiple awards for his teaching at UCSF, and election to the U.S. National Academy of Sciences and the Royal Society of London. He has published more than 400 research articles and the book How to Win the Nobel Prize: An Unexpected Life in Science.
What’s the Big Deal?
Cancer ranks among the most lethal and feared of human diseases. But until late in the 20th century, little was known about how the disease arises. Then, a remarkable cascade of discoveries uncovered the deepest secret of the cancer cell – malfunctioning genes. The malfunction takes two forms: jammed accelerators and defective brakes, which act in concert to create the malignant phenotype. Both sorts of culprits arise from normal genes by mutation or other disturbances. The normal precursors to jammed accelerators are known collectively as “proto-oncogenes,” the precursors to defective brakes as “tumor suppressor genes.” Together, these genes represent a genetic keyboard on which all manner of tumorigenic agents can play. Genomic science has now compiled an extensive inventory of the individual keys in this keyboard, providing an unprecedented opportunity to elucidate the functional maladies that lead to cancer. The “genetic paradigm” for cancer has had a profound impact on cancer research and inspired new approaches to the clinical care of cancer patients. A definitive conquest of the disease still eludes us, but we have an ever-deepening understanding of where the remaining obstacles lie.
Learning Overview
Big Concepts
Despite their diverse causes, all cancers ultimately arise from the malfunction of genes known as proto-oncogenes and tumor suppressor genes – the accelerators and brakes respectively that govern the lives of our cells. These genes represent a keyboard on which all sorts of carcinogens can play. Exploration of the keyboard is enhancing every aspect of our assault on cancer.
Bio-Dictionary Terms Used
allele, ATP, cDNA, carcinogen, cloning, chromosome, DNA complementarity, eukaryotic cell, gene, microtubule, mitosis, mutation, nucleus, pedigree, protein kinase, signaling pathways, substrate, virus
Terms and Concepts Explained
aneuploidy, chromosome translocation, molecular hybridization, oncogene, nuclease, proto-oncogene, reverse transcriptase, tumor suppressor gene, tyrosine kinase
Introduction
-
In 1909, Peyton Rous discovered a virus that causes cancer in chickens (now known as Rous Sarcoma Virus, or RSV). At first ignored by the scientific community, this discovery eventually sired a scientific revolution that produced a unifying genetic paradigm for all cancer, a paradigm that now guides both cancer research and the clinical care of patients with the disease.
Part I: Journey to Discovery – Normal Genes Can Be Perverted Into Oncogenes That Give Rise To Cancer
-
Cancer emerges when regulatory systems that keep cell division in check are broken and the cells divide in an uncontrolled manner.
-
As an example of such uncontrolled growth, cells derived from a cervical cancer from Henrietta Lacks in 1952 are still grown today in laboratories around the world (HeLa cells).
-
An early clue about the genesis of cancer came from Rudolph Virchow, who in 1858 noticed that cells from multiple metastases resemble one another, suggesting a common origin and, thus, a vast number of cell divisions.
-
Late in the 19th century, it became apparent that exposure to external agents such as noxious chemicals, sunlight, and X-rays can give rise to cancer.
-
Theodor Boveri in 1914 speculated that the gain or loss of chromosomes (aneuploidy) may give rise to cancer. In the 1960s, a specific chromosomal translocation (splicing of one chromosome arm with another chromosome) was found to be a hallmark of chronic myeloid leukemia.
-
In 1866, Pierre Paul Broca found that in certain families, cancer can be heritable from one generation to the next.
-
Investigation of the disease Xeroderma pigmentosum revealed that failure to repair DNA damage can give rise to cancer.
-
Mike Bishop decided that RSV had unique advantages as a means to study the genesis of cancer. He was soon joined by Harold Varmus as a research colleague in this project.
-
RSV has an RNA genome and a special enzyme called reverse transcriptase that copies the RNA into DNA, which in turn is required for replication of the virus. The discoverers of reverse transcriptase were awarded the Nobel Prize.
-
RSV has a gene called v-src that is responsible for tumor formation. The origin of v-src was a mystery. Bishop and Varmus decided to explore whether the gene might have originated from normal cells.
-
The scientists devised a DNA “probe” for detecting the presence of a cellular counterpart to v-src. This method (called “molecular hybridization”) involves a test-tube reaction between the single-stranded DNA probe and a complementary DNA strand in the cell genome.
-
The results of these experiments clearly showed that a gene closely related to v-src (now known as c-src) is present in both avian and mammalian genomes.
-
Later, v-src and c-src were discovered to encode a tyrosine kinase, an enzyme that transfers a phosphate group from ATP and a tyrosine amino acid on protein. This initiates a signaling process that tells the cell to divide. The kinase of v-src can phosphorylate numerous proteins, which provides an explanation for its ability to dramatically change the phenotype of a cell.
-
The difference between c-src and the pernicious v-src oncogene is a mutation in v-src that permanently turns on the kinase. The normal gene c-src version called a “proto-oncogene” while the pernicious v-src is called an “oncogene.”
Part II: Knowledge Overview – The Origin of Cancer
-
Cancer is characterized by abnormal cellular behaviors that promote the growth, survival, and metastasis of tumors. Examples include 1) continuous signals for cell division, 2) evading growth suppression, 3) resisting cell death, 4) immortalization, 5) invasive behavior, and 6) blood vessel formation (angiogenesis).
-
The complexity of the malignant phenotype was perplexing until it was traced to the malfunction of genes that control the behavior of cells.
-
One type of genetic malfunction results in a gain-of-function (a dominant trait), analogous to “jammed accelerators” for division. These malfunctioning “oncogenes” can arise from 1) changes to the nucleic acid sequence of genes, 2) chromosome translocations, 3) amplifications that increase the copy number of a gene, or 4) malfunctioning control of their expression by elements external to the gene.
-
Another type of genetic mishap causes a loss-of-function (a recessive trait), analogous to a “defective brake.” One example of a tumor suppressor is the retinoblastoma gene RB1; when both copies are lost or defective, then retinoblastoma will likely arise. BRACA1 and 2 are other examples of tumor suppressor genes, defects which are associated with breast cancer.
-
Cancer cells also frequently have abnormalities of chromosomal number and structure, including missing or extra copies of chromosomes (aneuploidy), deletions and amplifications within chromosomes, or translocations between chromosomes.
-
At least 25% of human cancers are caused by viruses. Examples discussed in the text are Epstein Barr Virus (EBV, Burkitt’s lymphoma and several other tumors), Hepatitis B Virus (HBV, liver cancer), Hepatitis C Virus (HCV, liver cancer), and Human Papilloma Virus (HPV, cervical cancer and several other tumors).
-
Vaccines have been successfully made against HBV and HPV, which provide a means for preventing cancer caused by these viruses.
-
Tumors appear to arise in a stepwise manner. Cancer begins with a single cell that suffers an initial genetic malfunction and proceeds through multiple subsequent genetic alterations, each one providing greater survival value to the emerging cancer cell. For this reason, the risk of most cancers increases with age.
-
An international consortium has compiled the complete nucleotide sequences of a vast number of human cancer genomes. Cancer genomics is identifying mutations in “driver genes” that tumorigenesis and “passenger” mutations that represent innocuous collateral damage from whatever created the driver genes.
-
Mutations in cancer cells can be traced to external agents (UV light or environmental substances such as smoke) as well as mutations that arise spontaneously.
-
Some individuals have heritable risk of cancer, examples being found in the retinoblastoma and BRCA genes.
-
Early detection of cancer is likely to increase the success of therapy. There are screening tests for breast, cervix, colon, lung, and prostate cancer, and new, highly sensitive genomic tests are being developed.
-
Genomic test can also be used for prognosis of cancer patients.
Part III: Frontiers – Exploiting The Cancer Genome For Therapy
-
Targeted therapies for cancer are being developed. One approach is to inhibit or otherwise target the “jammed accelerators” (oncogenes). Another approach is coaxing the immune system to kill cancer cells (cancer immunotherapy).
-
Chronic myelogenous leukemia (CML) is a blood cancer that arises from a chromosomal translocation that produces a hyperactive state (jammed accelerator) of the Abl tyrosine kinase, resulting in uncontrolled cell division.
-
A chemical inhibitor of Abl was identified (Gleevec), which has resulted in a very successful drug treatment for CML patients.
-
Through mutations in the Abl gene, cancer cells can become resistant to Gleevec, but pharmaceutical companies have developed new drugs that work against mutated Abl.
-
Gleevec is potentially a lifelong therapy because it fails to completely eradicate malignant cells, leaving a reservoir that leads to recurrence if drug treatment is terminated.
-
Pharmaceutical companies developed inhibitors of the EGF receptor kinase for lung cancer, but they failed initial clinical trials.
-
A subset of lung cancer patients (tending to be young, Asian, nonsmokers), however, responded well to the EGF receptor inhibitors. DNA sequencing revealed that they had a unique genetic signature – a mutation in their EGR receptor gene. Subsequent clinical trials with this patient subgroup produced a positive clinical result.
-
Patients with non-small cell lung cancer are now routinely screened for EGR receptor gene mutations. This is an example of what could be more prevalent in the future – personalized therapy based upon the genetic signature of a patient’s cancer.
Closing Thoughts
-
The ability to rapidly sequence genomes at a relatively low cost has greatly accelerated the current revolution in cancer research, which has produced a genetic paradigm for the disease.
-
Rous left two path-breaking legacies: the discovery that viruses can cause cancer, which inspired the eventual identification of viruses that cause human cancer; and a chain of research that eventually linked wayward genes to cancer.
-
The conviction that study of a chicken virus might lead to discoveries that are applicable to human cancer was based on a faith in the universality of nature, a faith that pervades modern biological research and has returned numerous advances.
Guided Paper
Stehelin, D., Varmus, H.E., Bishop, J.M., and Vogt, P.K. (1976). DNA related to the transforming virus gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature, 260: 170–173.
This paper demonstrates that the DNA of normal chicken cells contains DNA sequences closely related to the genes of an avian cancer-causing virus. This knowledge reveals the origins of the genomes of such virus and the roles of these genes in cancer. This research led to the Nobel prize being awarded to Bishop and Varmus in 1989.
DownloadIntroduction
On May 14, 2013, the film star Angelina Jolie published an article in the New York Times, not on her latest film, but rather explaining her decision to undergo a bilateral mastectomy. Her decision was not prompted by a cancer diagnosis but by the results of a genetic test that revealed that Ms. Jolie was likely (>80% chance) to develop breast cancer in the future, if the mastectomy was not performed. If a breast cancer were to arise, its successful treatment could not be guaranteed. She later had her ovaries removed as well because they too were at high risk of developing cancer. Ms. Jolie’s brave choice of making her story and decision public dramatized an important aspect of cancer – that it is fundamentally a genetic disease. Sometimes faulty genes are acquired through families, as was true for Ms. Jolie whose mother died of breast cancer at age 56, but most often defects in genes arise through insults to our DNA as we age.
This Narrative outlines how we came to know of cancer as a genetic disease, and how such knowledge has transformed both cancer research and the clinical care of cancer patients. These advances in turn have raised new hope that we might eventually succeed in our efforts to control and perhaps even eradicate cancer. The Narrative is rich with illustrations of how science proceeds, full of surprises, times when path-breaking results are not readily accepted by the research community, and examples of how scientists build upon one another’s work to come to new insights.
The story that I will tell begins humbly enough in 1909, when a farmer on Long Island, New York, noticed a tumor on the breast of one of his Plymouth Rock hens. He watched the tumor grow for two months and then took the bird to the Rockefeller Institute in New York City to see what could be done.
The farmer was referred to Peyton Rous, a young pathologist with an interest in cancer (although he had never performed any research on the subject). Rous is said to have been a forceful, even fiery individual. Perhaps by brunt of his personality, he convinced the farmer to donate his chicken to medical research. Instead of attempting to cure the chicken, Rous used it to make a path-breaking discovery: the chicken’s tumor contained an infectious agent that could cause cancer.
Rous first showed that a tumor can be transplanted from one chicken to another and it will continue to grow. More importantly, he then found that cell-free extracts of tumor cells induced a similar tumor when injected into a chicken (Figure 1). It seemed likely that the ability of the extracts to elicit cancer resided in an infectious agent much smaller than a bacterium. Rous announced this discovery with a two-page article in – of all places – the Journal of the American Medical Association. You will not find much about chickens in that journal in this day and age. The infectious agent was a virus that was later named the Rous Sarcoma Virus (or RSV) in honor of the man who first isolated it and the malady that it created in the chicken (sarcoma is a solid tumor of muscle and connective tissue, and relatively rare in humans). This virus is a key actor in a nearly century-long story that I will unfold in the Journey to Discovery.
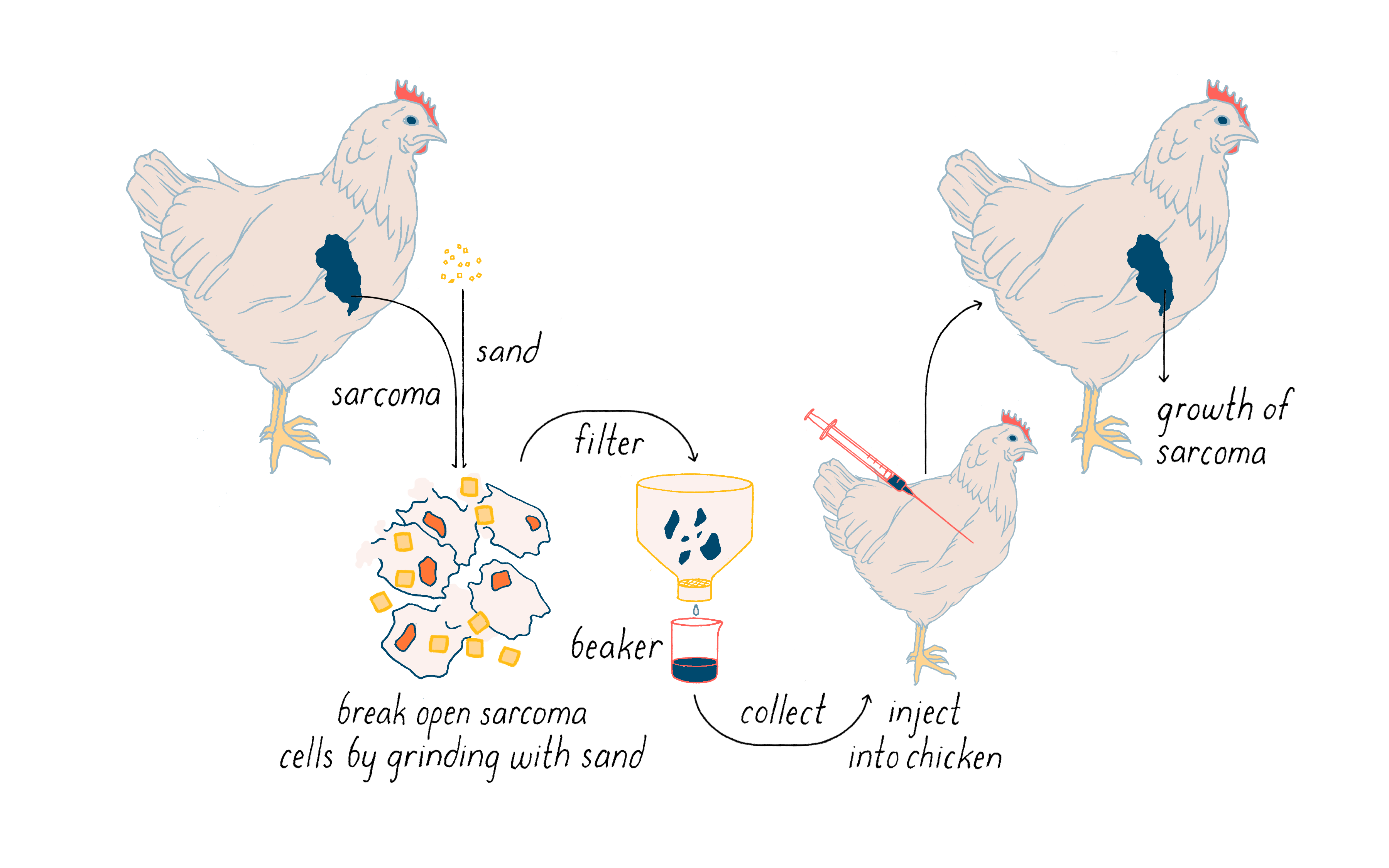
The work with chicken viruses was greeted with what Rous described as “downright disbelief,” and few scientists showed any interest in the finding. Rous himself eventually became disillusioned when he failed to identify causative viruses in transplantable tumors of rodents. It would be 55 years before his discovery of the chicken tumor virus earned Peyton Rous the Nobel Prize at age 85. Rous opened his Nobel Lecture in the following manner:
“Tumors destroy man in a unique and appalling way, as flesh of his own flesh which has somehow been rendered proliferative, rampant, predatory and ungovernable. [Tumors] are the most concrete and formidable of human maladies, yet despite more than 70 years of experimental study, they remain the least understood… What can be the why for these happenings?”
We now know the why for these happenings, and it takes the form of a unifying principle. No matter what its causes – and there are many of those – cancer ultimately arises from the malfunction of genes. This was the insight first glimpsed by Harold Varmus and myself, using the Rous Sarcoma Virus as a tool. In the Knowledge Overview, I will cover the different ways in which genes can malfunction so as to give rise to cancers. One type of malfunction can be equated to jammed accelerators, which represent a large number of genes known as “proto-oncogenes.” Another malfunction is comparable to defective brakes, affecting genes known as “tumor suppressor” genes. In a nasty collaboration, the two forms combine to create malignant cells that maim and kill. As part of this exploration, I will also describe the specific genes (BRCA1 and 2) that accounted for the untimely death of Ms. Jolie’s mother and were the subject of the genetic testing that prompted Ms. Jolie’s drastic action. I will describe the broad impact that our knowledge of cancer genetics is having on our effort to conquer cancer. And in the Frontiers section, I will explore in some detail how the genetic paradigm for cancer is influencing the development of new therapeutics for cancer.
Part I: Journey to Discovery —
Normal Genes Can Be Perverted into Oncogenes That Give Rise to Cancer
The Problem
Cancer is a disease of cells, the microscopic bricks with which all creatures are built. Cells make their own fuel, converse with each other by means of molecular signals, and perform other marvelous functions. They reproduce themselves as necessary – ten thousand trillion times during the course of a human life time.
Cancer emerges when cell division has gone awry. Most cells in the adult human body have stopped dividing or divide occasionally to replenish tissues or in response to injury. Cells take stock of the cues in their environment before deciding to divide. Cancer cells, however, have stopped listening. The regulatory systems that keep cell division in check are broken and the cells divide in an uncontrolled manner, piling up unwanted cells in the blood or in a tumor, which can eventually, and sometimes rapidly, lead to death.
The uncontrolled growth of cancer cells can continue even past the death of a patient, if the cells are attended to and fed. Consider HeLa cells, human cancer cells that are grown in culture for research. In 1952, Henrietta Lacks was treated for cervical cancer at Johns Hopkins University. Without her knowledge, cells from her tumor were placed into tissue culture, where they have thrived to this day and are widely used in medical research around the globe. Henrietta Lacks and her immortal remnants have become a cause célèbre in the United States, because of a book about them that was on the New York Times bestseller list for several years [The Immortal Life of Henrietta Lacks, by Rebecca Skloot].
According to sources cited in Wikipedia, HeLa cells (shown in Figure 2) have now grown to a cumulative bulk of 50 million metric tons (and still increasing). They remain highly malignant and show no sign of slowing down their replication. The extraordinary stability of the malignant state in HeLa cells hints at a genetic underpinning.

Almost everyone knows of someone who has died of cancer. Cancer is neck-in-neck with heart disease for the leading cause of death in the United States, accounting for ~600,000 deaths per year. However, progress is being made. We knew so little about cancer a few decades ago that the only treatments other than surgery were “cell poisons” that have only marginal differences in killing cancer versus healthy cells. Now, we have more specific therapies that have come from knowledge that the DNA in cancer cells is not identical to that in healthy cells.
What follows is the story of how we came to this fundamental understanding of cancer. The story begins with a series of clues that gradually accumulated over the course of the last century, only to be ignored until the virus discovered by Peyton Rous led us to the heart of the cancer cell – its genome.
Clues
Clue 1: Cancers arise from a clonal origin and then multiply
The earliest and most fundamental clue was the fact that malignancy is a highly durable property (or “phenotype,” as geneticists would say). In 1858, the German pathologist Rudolf Virchow noticed that the cells of multiple metastases in an individual all resemble one another, as well as those of the primary tumor. He concluded that all these cells might have a common origin, which in turn implied a large number of cellular divisions, throughout which the malignant phenotype had been preserved.
At the time, it was widely believed that every cell was newly created out of a mythical substance known as the cytoblastema. Virchow’s observations with cancer cells persuaded him that a newly emerging and opposing theory was correct: all cells come from cells. He became the theory’s most ardent and effective advocate, to the point of being mistakenly credited with originating the theory himself.
Virchow’s inference about the common origin of cells in a primary tumor and its metastases inadvertently foreshadowed a modern tenet of tumorigenesis. Cancers originate from single wayward cells whose astonishing fecundity engenders the billions of cells that finally compose a malignant tumor. In medical terms, a tumor is typically “monoclonal,” arising from a single progenitor cell. However, a lot happens to the multiplying cells as the tumor emerges – I will return to that later.
Clue 2: Mutations give rise to cancer
The second clue was the discovery that many causes of cancer damage DNA, creating what geneticists call “mutations” (see also the Narrative on Mutations by Koshland).
The path to this discovery had two phases.
The first of these was the realization that many cancers are caused by agents originating from outside the body.
The notion that external agents can cause cancer emerged in England in 1761 when the London physician John Hill reported that the inhalation of snuff (or smokeless tobacco in the modern, sanitized term) was associated with the occurrence of nasal cancer. Fourteen years later, Percival Pott reported that the chimney sweeps of Britain (see Figure 3) were highly prone to the otherwise rare cancer of the scrotum. He attributed this to chimney soot. French chimney sweeps were reported to be less afflicted. By way of explanation, Pott noted that the French of his time bathed more frequently than the Brits.

The industrial revolution of the late 19th century exposed humans to large quantities of noxious agents, some of which proved to be vigorous carcinogens. At roughly the same time, it was noticed that individuals who spent an exceptional amount of time in the sun were unusually prone to skin cancer.
Soon after the discovery of X-rays, individuals who had been working with this new-found form of radiation began to develop malignant tumors at sites of exposure. To this point, the evidence was limited to guilt by association. The cancers of the early workers with X-rays then inspired the Parisian physician Jean Clunet to do an experiment. Clunet irradiated four white rats. Two died immediately, the third developed an invasive sarcoma at the site of irradiation after a hiatus of more than 12 months; the fourth remains unaccounted for. However frail the experiment now seems, it was published and is still viewed as the first induction of cancer in an experimental animal.
Seven years later, Katsusaburo Yamagiwa in Japan repeatedly applied coal tar to the ears of rabbits and obtained malignant tumors of the skin. Ernest Kennaway in Britain then refined Yamagiwa’s discovery by purifying a carcinogenic activity from coal tar, identifying it as benzanthracene, and then synthesizing it from scratch and demonstrating its carcinogenicity. Yamagiwa was so pleased with his discovery that he wrote a haiku. Roughly translated, it reads
“Cancer was produced, Proudly, I walk a few steps.”
The threads all came together in 1981, when the British epidemiologists Richard Doll and Richard Peto published a magisterial study of what might cause human cancer. They concluded that at least 80% of human cancers are due to external factors and were thus, in principle, preventable. This number may be at least a modest overestimate because we now know that many of the mutations in cancer cells originate from spontaneous mistakes by the machinery that replicates our DNA (see the Narrative on Mutations by Koshland). But the importance of external causes of cancer remains unchallenged and, for some cancers, demonstrable by genomic analysis (see the Knowledge Overview).
The discovery of external carcinogens immediately raised the question of how they might actually work. The first insight came in 1926 from H. J. Muller, when he demonstrated that X-rays caused mutations in fruit flies. He concluded his first report of this finding with the suggestion that mutation might explain the cancers elicited by radiation. The thought fell on deaf ears.
Within a decade of Muller’s discovery, however, the carcinogen methylcholanthrene was shown to cause mutations in mice. Much later, in the early 1970s, Bruce Ames and others demonstrated that many (but far from all) chemical carcinogens are mutagenic when applied to bacteria, which gave rise to a test for potential carcinogens (see also the Narrative on Mutations by Koshland).
When all was said and done, however, none of this work produced a consensus that mutation of genes might be responsible for cancer.
Clue 3: Cancers have abnormalities in chromosomes
Meanwhile, a third clue was emerging. Cancer cells have both numerical and structural abnormalities of chromosomes.
This story begins with the German biologist Theodor Boveri, who spent his career using sea urchins and worms to study cells and chromosomes. From this work, Boveri was able to show that chromosomes are the physical carriers of heredity. Working as a graduate student, the American William Sutton reached the same conclusion at the same time, using experiments with grasshoppers.
Boveri also sometimes observed abnormal arrangements of chromosomes in spindles, which he recorded in drawings such as Figure 4 (see the Bio-Dictionary video for a short description of the normal process of mitosis). Then, in a stunning feat of imagination, without ever laying hands on a cancer cell, Boveri conceived the idea that gain and loss of individual chromosomes might be responsible for cancer – a circumstance that we now call aneuploidy and known to be common in cancer cells. He developed this idea in a monograph that was published in 1914, which went unappreciated at the time, but is now considered one of the great classics of the medical literature.
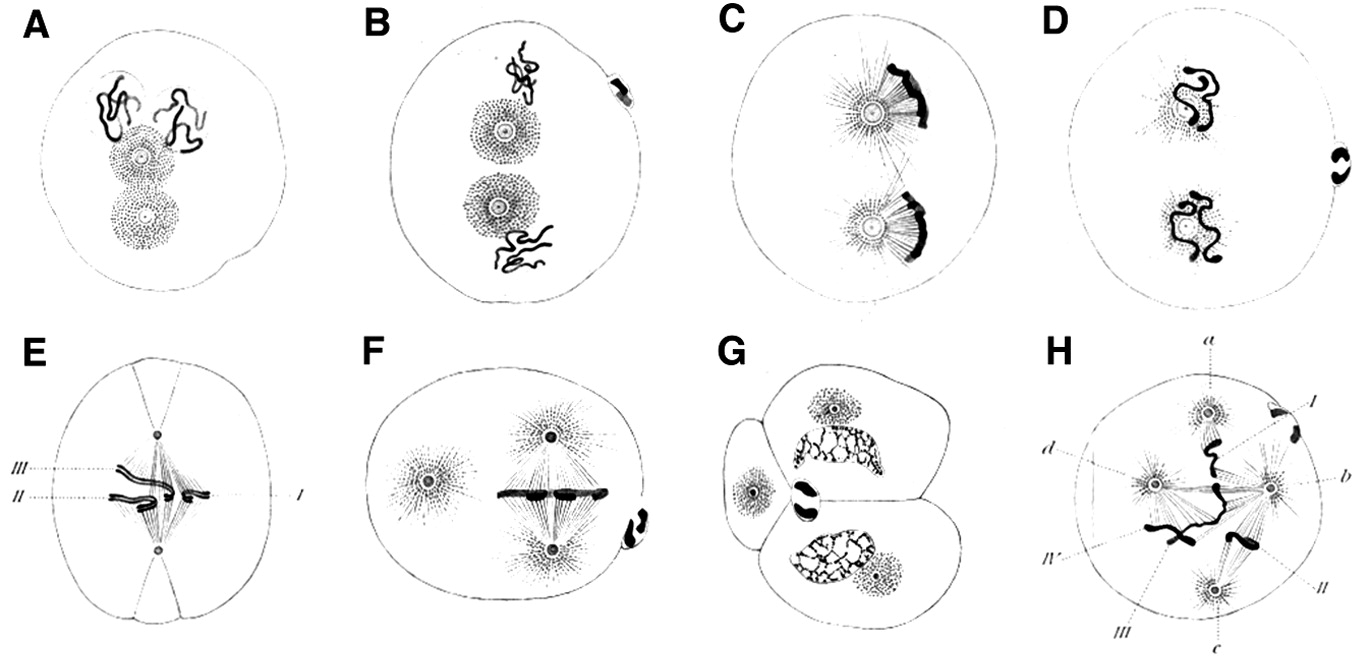
Boveri’s theoretical incrimination of chromosomes in cancer lay fallow until 1961, when David Hungerford and Peter Nowell uncovered a miniature chromosome that was consistently present in the cells of chronic myeloid leukemia (CML) – the Philadelphia Chromosome, named for the city in which it was discovered. It is worth noting that Hungerford was a mere graduate student at the time, but it was his skill at examining chromosomes that made the discovery possible. Janet Rowley would eventually show that the miniature chromosome is the product of a reciprocal translocation between chromosomes 9 and 22 – the two chromosomes swap pieces of themselves, creating abnormal chromosomes. These remarkable discoveries ultimately gave rise to a dramatic therapeutic advance – the wonder drug Gleevec, which has transformed the treatment of the disease in which the Philadelphia Chromosome was first found. This story will be covered in the Frontiers section.
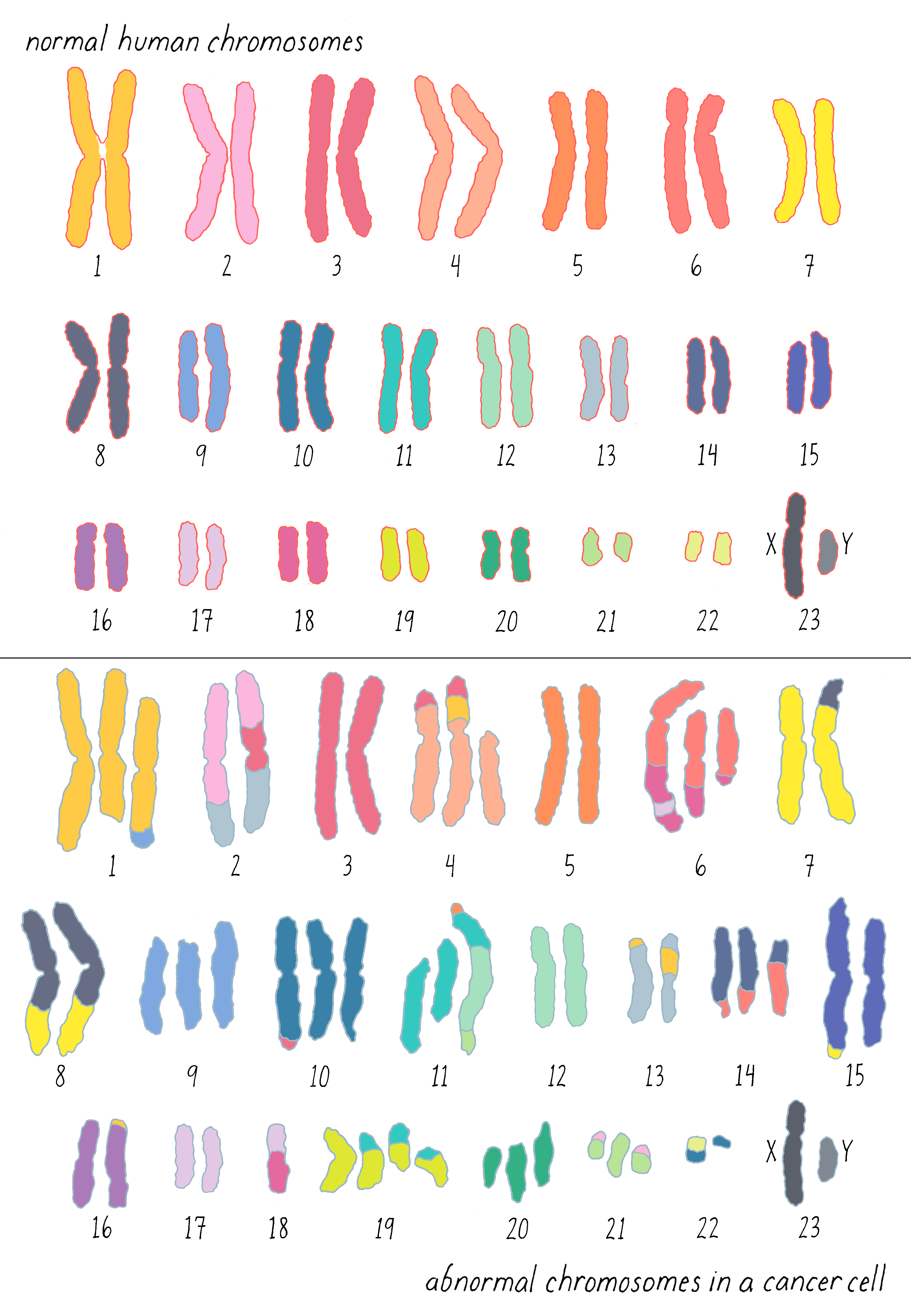
Gradually, translocations in other forms of leukemia came into view. For a while, chromosomal translocations were thought to be exclusive to leukemias, but that proved to be a misapprehension created by the relative difficulty in studying the chromosomes of solid tumors. Soon after it became apparent that both chromosomal number and structure are present in virtually all malignancies, as exemplified by the chromosomes of a colon cancer shown here (Figure 5): it is sheer mayhem, and it is easy to imagine that this mayhem might disturb the behavior of cells.
Clue 4: Cancer run in certain families
The fourth clue strikes at the heart of the matter: cancer is sometimes heritable.
In 1866, the neurosurgeon and anthropologist Pierre Paul Broca took time out to sketch the medical pedigree of his wife’s family and found himself confronted with an inherited predisposition to breast cancer: 9 of the 25 women in the family tree had developed the disease, as shown in Figure 6.
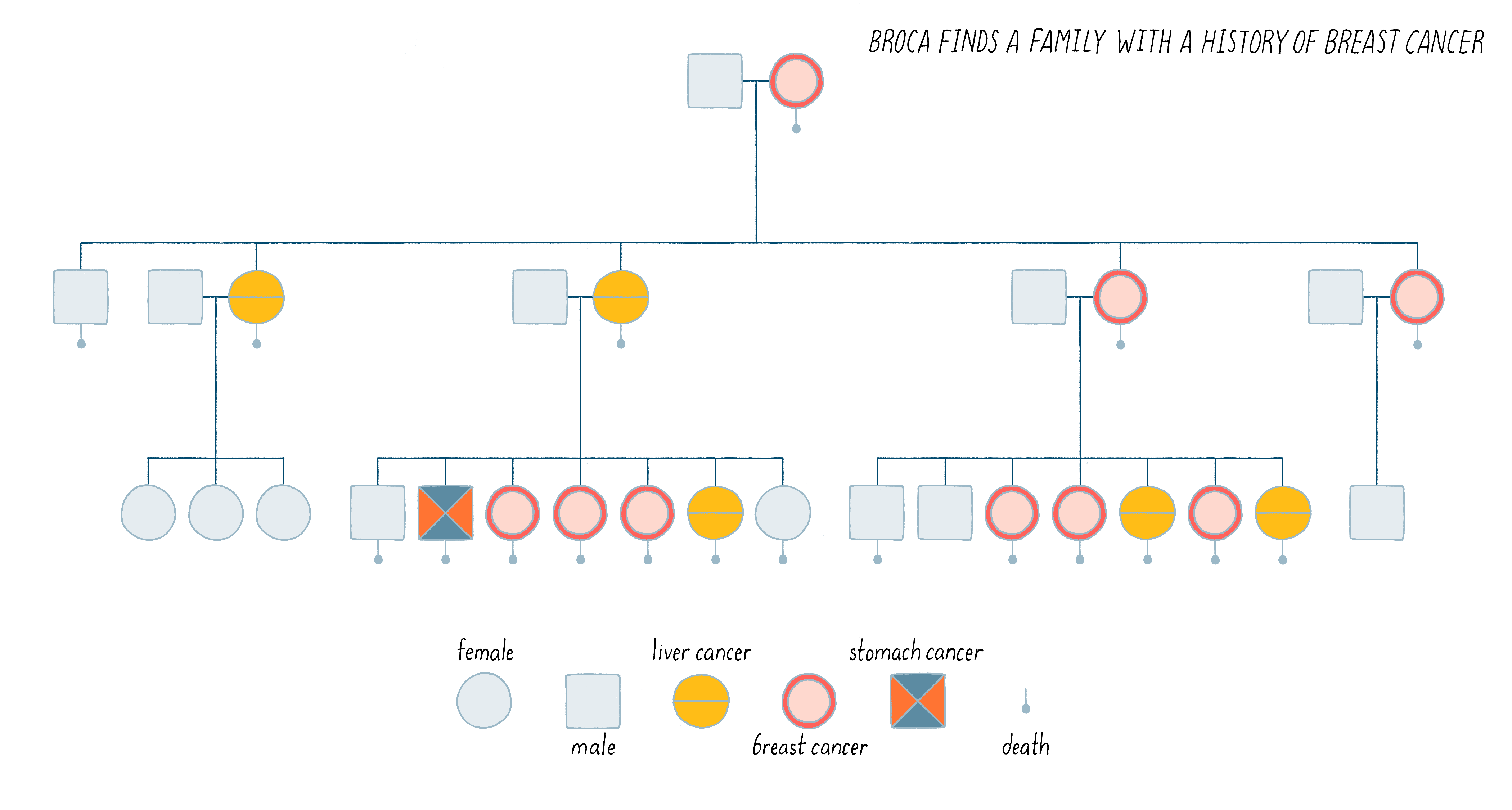
Broca went on to expand this observation by identifying multiple human pedigrees in which cancer seemed to be inherited. It was an insight far ahead of its time. We now know that perhaps 10% of human cancer is familial, inherited in the manner described by Broca. Indeed, this is the case for Angelina Jolie’s family described in the Introduction. We will discuss the nature of her family’s heritable disease in the Knowledge Overview.
Clue 5: Failure to repair DNA is associated with cancer
The fifth and final clue involved the ability of our cells to repair damage to their DNA. Maintaining the integrity of DNA is a vital issue for cells, so evolution has provided multiple and distinct mechanisms for the repair of damaged DNA. Inherited defects in that machinery make the cellular DNA more vulnerable to mutations and thus can create a predisposition to cancer.
The first example came from the study of patients with Xeroderma pigmentosum, a horrendous disease in which the UV rays in sunlight induce skin cancers in afflicted individuals. In 1968, James Cleaver reported that individuals with Xeroderma pigmentosum have inherited a defect in the machinery that normally repairs the DNA damage caused by UV light. It was an easy leap to suspect that unrepaired DNA might be a seedbed for cancer. In the ensuing years, other defects in the response to DNA damage have been implicated in both heritable and somatic cancers (Angelina Jolie’s “bad” gene among them).
The Discovery- A Cellular Origin of Cancer-Causing Genes
The diverse clues were all in hand by the late 1960s. But many still turned a blind eye, for want of direct experimental evidence that genes can cause cancer. Waiting in the wings to address that deficiency was RSV, which brings us to my part of the story.
I first encountered the Rous Sarcoma Virus when I arrived at UCSF in 1968, introduced to it by Warren Levinson, another new member of the UCSF faculty. Here is what caught my attention: infection of chicken cells with the virus converts them to a facsimile of cancer cells within 24 hours (a process in the cancer field referred to as “transformation”; Figure 7). Picking a good experimental system is critical for solving problems in biology. It seemed to me that the virus of Peyton Rous represented a promising handle on the door to the mysteries of cancer.

Explorer’s Question: What made Rous Sarcoma Virus a good system for studying cancer?
A. It was a simple and reproducible means of rapidly converting cells from normal to cancerous growth.
B. There was a quantitative assay for the virus in vitro, which made the virus amenable to genetic analysis.
C. The virus could be purified in large quantities, facilitating physical and biochemical analysis.
D. And it was safe to use – it does not infect human cells.
Answer: All of the above are correct. Our motivation was to understand the basic principles underlying the tumorigenicity of a chicken virus (points A–D were useful for our studies). However, we also reasoned that studying a chicken tumor virus would provide insight into the origins of cancers in humans as well, which indeed proved to be correct.
At the time, Rous Sarcoma Virus posed two great puzzles.
1) It appeared that the viral genome could be established as a heritable property of the host cell. Therein lay a puzzle. The genome of RSV consists of RNA, not DNA. Biologists knew, beginning with Watson and Crick, how DNA was replicated and inherited (see the Narrative by Vale on DNA Structure), but there was no known way for RNA to be inherited in the manner of a gene made of DNA.

2) The Rous Sarcoma Virus was capable of transforming normal cells into cancerous growth. How is that accomplished?
The first puzzle was solved in the late spring of 1970 when Howard Temin and David Baltimore independently discovered an RNA-directed DNA polymerase within RNA tumor viruses, which soon acquired the name “reverse transcriptase.” (In turn, RNA tumor viruses were rechristened “retroviruses”; HIV is a particularly noxious retrovirus.)
Their discovery and how reverse transcription allows retroviruses to replicate is described in Video 2.
The discovery of reverse transcriptase was a transformative event intellectually. Reverse transcription modifies the unidirectional arrow of the Central Dogma championed by Francis Crick in which genetic information can flow in only one direction: from DNA to RNA and then on to protein. Thanks to the enzyme reverse transcriptase, RNA can be copied into DNA (Figure 9), thus violating the Central Dogma by reversing the flow of genetic information. The ability to copy any RNA into DNA with reverse transcriptase was invaluable to both fundamental research and the burgeoning biotechnology industry; cDNA cloning, for example, relies on reverse transcription. For my colleagues and me, reverse transcription was a Godsend for our work, as you will soon see.

The second puzzle – how does the Rous Sarcoma Virus cause cancer? – was the basis of a 16-year collaboration between myself and Harold Varmus. Harold joined me in 1970, following a postdoctoral fellowship at the National Institutes of Health. He soon became my coequal in directing the research in our laboratory, rose quickly through the ranks of the faculty, and later served first as the Director of the National Institutes of Health and then as the President of the Memorial Sloan Kettering Cancer Center, and most recently, as the Director of the National Cancer Institute.
The genome of Rous Sarcoma Virus is relatively simple, containing only 4 genes encoding 6 proteins (see Dig Deeper 1 for description of the RSV genome). One of the genes is called v-src (v for “viral”), and it plays no role in viral replication or construction. Genetic analysis revealed that v-src was an “oncogene,” meaning that it was responsible for malignant transformation of host cells (src is short for “sarcoma,” the type of tumor elicited by the virus).
The gene v-src raised an evolutionary puzzle. If it is irrelevant to viral replication, why is it there? Two opposing lines of thought directed us to the genome of normal cells.
First, there was the “virogene-oncogene” hypothesis formulated by Robert Huebner and George Todaro. The thought was that retroviruses had deposited themselves with their oncogenes in the germlines of ancient ancestors of modern species. Normally repressed by the host cell, these genes might be activated by various carcinogenic agents. By this account, we might possibly find a src gene in normal cells (termed c-src for “cellular src”), but it would be in the form of a retroviral oncogene, and its origin would remain unexplained.
The opposing thought was Darwinian in nature. Since v-src is irrelevant to the viral life cycle, it is not likely to have arisen in concert with the remainder of the viral genome – there was no apparent selective pressure to accomplish that. Instead, the intimate interaction between retroviral and cellular genomes might have created an accident in which a cellular gene (c-src) was incorporated into a retroviral genome – in essence, reversing the hypothesis of Huebner and Todaro. This thought also favored a search for src in normal DNA.
To explore these two ideas, we had to devise a method for detecting a gene related to v-src in normal chicken cells with great sensitivity and specificity. Our general strategy was to make a single-stranded DNA “probe” of v-src. This single-stranded viral probe could react with a complementary single-stranded DNA from cellular src (c-src) if it existed to form a double-stranded DNA through Watson-Crick base pairing (Figure 10). This test-tube reaction between one single-stranded DNA (or RNA) and a partner complementary strand (either DNA or RNA) is called “molecular hybridization.”

Making the single-stranded DNA “probe” for v-src was a laborious effort, mainly because we did not have the benefit of recombinant DNA, which was not yet available. We made do with the tools at hand.
At our disposal was a mutant form of the Rous Sarcoma Virus that lacks most of src (found by our collaborator Peter Vogt). We call this RSV-td, “td” meaning transformation deficient. This unique reagent was vital to our experiments – there was nothing like it then available for any other retrovirus – and Peter joined us as a coauthor on our initial report of the results with the probe.
As shown in Figure 11, we could use the RNA of a “td” mutant to perform what later came to be called “subtractive hybridization.” This is where the reverse transcriptase discovered by Baltimore and Temin also provided a critical tool that made our study possible. Here were the various key steps:
1. Reverse transcribe (copy) the genome of RSV into cDNA in a test tube. During this copying, radioactive nucleotides were introduced which became incorporated into the cDNA. The radioactivity would allow us to track this cDNA in a later experiment.
2. cDNA is single-stranded and it has the potential to form a double-helix with a partner strand that has complementary matching nucleotides via Watson-Crick base pairing. Hybridize the cDNA to RNA from the deletion mutant, which would capture all DNA except that representing v-src.
3. Remove the double-stranded hybrids, leaving the single-stranded, radioactively labeled cDNA for v-src – the probe we needed.

The radioactive v-src probe was not the final experimental result, but rather a tool for our critical experiment described next. With this radioactive cDNA of v-src, we could now probe whether there was a similar gene lurking in the genome of the chicken and other birds. In our experiment, we performed the following steps (see Figure 12):
1) Prepare DNA from the birds and then heat it to near boiling temperatures, which causes double-stranded DNA to dissociate into single strands.
2) Add our radioactive v-src single-stranded DNA to the heated, single-strand bird DNA, with the latter in great excess of the radioactive probe in order to drive the hybridization reaction. Then, the temperature was reduced to 68°C for several hours. At this temperature, two single DNA strands can come together to form a stable double helix, but only if there is a high degree of complementarity between the bases of the two strands. In this step, the v-src cDNA could hybridize with one strand of c-src gene in the bird genome, if such a gene existed. If such a gene did not exist, then the v-src cDNA would remain single-stranded.
3) After the above hybridization step, a nuclease specific to single-stranded DNA was added. This enzyme would chew up single-stranded DNA but leave double-stranded DNA untouched. Thus, the only way in which our v-src cDNA could survive is if it managed to find a partner strand in the cellular genome. We could test this by determining if the radioactivity in our viral src cDNA remained as intact DNA or whether it was degraded to single nucleotides by the added nuclease.
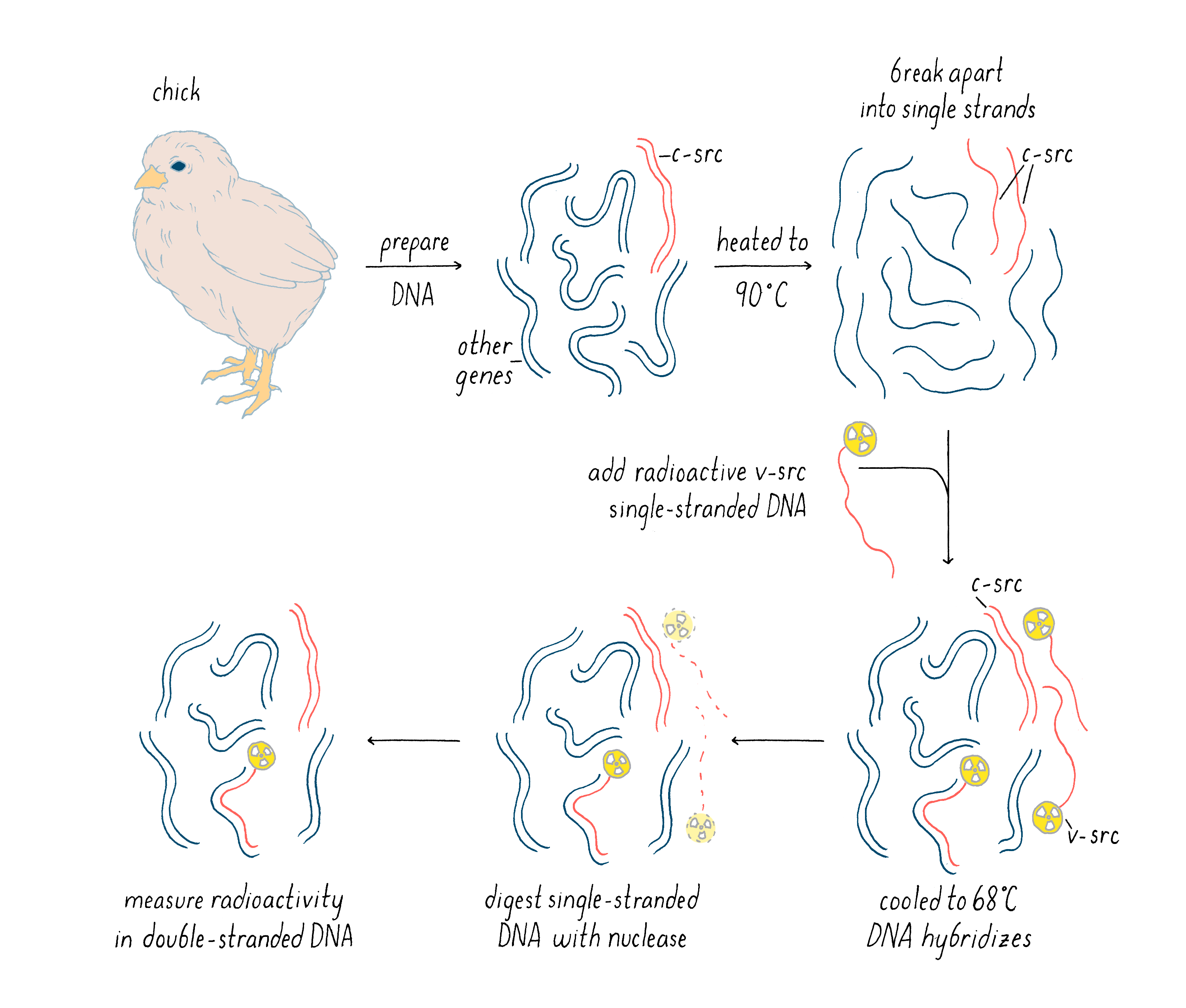
Explorer’s Question: When the DNA is cooled down in this experiment, the single-strand of one c-src gene can hybridize with the other strand of c-src (as before it was heated). How then could the hybridization of v-src to c-src be detected?
Answer: Yes, the two DNA strands of the cellular src gene could reanneal as well. However, the cellular DNA is in vast excess to the radioactive v-src cDNA and some of the c-src will find a v-src cDNA instead of itself. MANUAL
With this experiment, we had a persuasive answer to our question: the v-src probe hybridized with a close match to DNA in the natural genome of the chicken. We also found c-src in other birds such as quail, duck, and emu (a more ancient bird). This result was a game-changer: it suggested that the viral oncogene had been pirated from a host cell, and the conservation during the course of avian evolution made it likely that the cellular counterpart to v-src was indeed an essential cellular gene (c-src), rather than a wayward viral gene. Once recombinant DNA became available, we were able to isolate the c-src gene and show that its structure was indeed that of a normal cellular gene.
We published these findings as a brief note in the leading scientific journal Nature: 2 figures and 2 tables. The simplicity of our paper belied the extraordinary difficulty of the underlying experiments and the stunned reaction of the research community. We were pleased with ourselves.
Oh, how times have changed. Recently, a colleague told me that he had asked a class of graduate students to read that paper. Their reaction: “They got the Nobel Prize for that?”
I understand their reaction from a technical standpoint but wonder whether they grasped the impact of the result at the time. Today, the work could probably be done in a matter of weeks. When we did it, the experiments occupied more than two years. The fact that they were done at all was a tribute to the valiant efforts of two postdoctoral fellows, Ramareddy Guntaka and Dominique Stehelin. After two years of effort, the first sighting of a gene related to v-src in normal DNA was a riveting event. Here is how Dominique Stehelin later described his reaction:
“The fantastic results came out in the night of Saturday, October 6th, 1974: normal DNA contained sequences related to the src gene of the transforming virus … I suspect that few have had the privilege of enjoying such a moment when one is intensely and profoundly aware that a major step forward in Science has been made, and that one has contributed to it.”
I like to use that quote whenever I am trying to convince young people that doing science can be exciting – there is nothing quite like a moment of discovery.
After discovering c-src in bird genomes, we went on to show that the gene is also found in the mammalian genome, including that of humans. It was now clear that we had uncovered a gene with an ancient origin that, given its strong evolutionary conservation, likely serves a vital function for normal cells.
What Happened Next?
The discovery of c-src raised obvious questions. First, what is the function of the protein encoded by the two forms of src? Second, what is the difference between the well-behaved c-src and the mischievous v-src – how can we account for the malevolence of the latter? And third, is c-src unique in its ability to give rise to a cancer gene, or are there other such genes in normal cells?
In 1978, our group, as well as Ray Erikson’s lab, discovered that v-src encodes a protein kinase, an enzyme that extracts a phosphate group from ATP and transfers it to an amino acid of protein. At the time of this discovery, the only protein kinases known in eukaryotic cells transferred phosphates to serines or threonines. But in 1980, Tony Hunter discovered that the protein encoded by v-src was a tyrosine kinase, meaning that it transferred phosphate to a tyrosine (Figure 13). The same soon proved true for the c-src protein. Hunter’s result for v-src was followed by the uncovering of many more tyrosine kinases in later years. The human genome has nearly 100 tyrosine kinase genes. These tyrosine kinases are key “switches” that control the flow through signaling pathways in cells, including those that govern cell growth and division. Moreover, many are promiscuous (including the src kinase), phosphorylating numerous proteins. Given that promiscuity, it was easy to imagine how the unleashed v-src kinase might elicit the malignant phenotype.
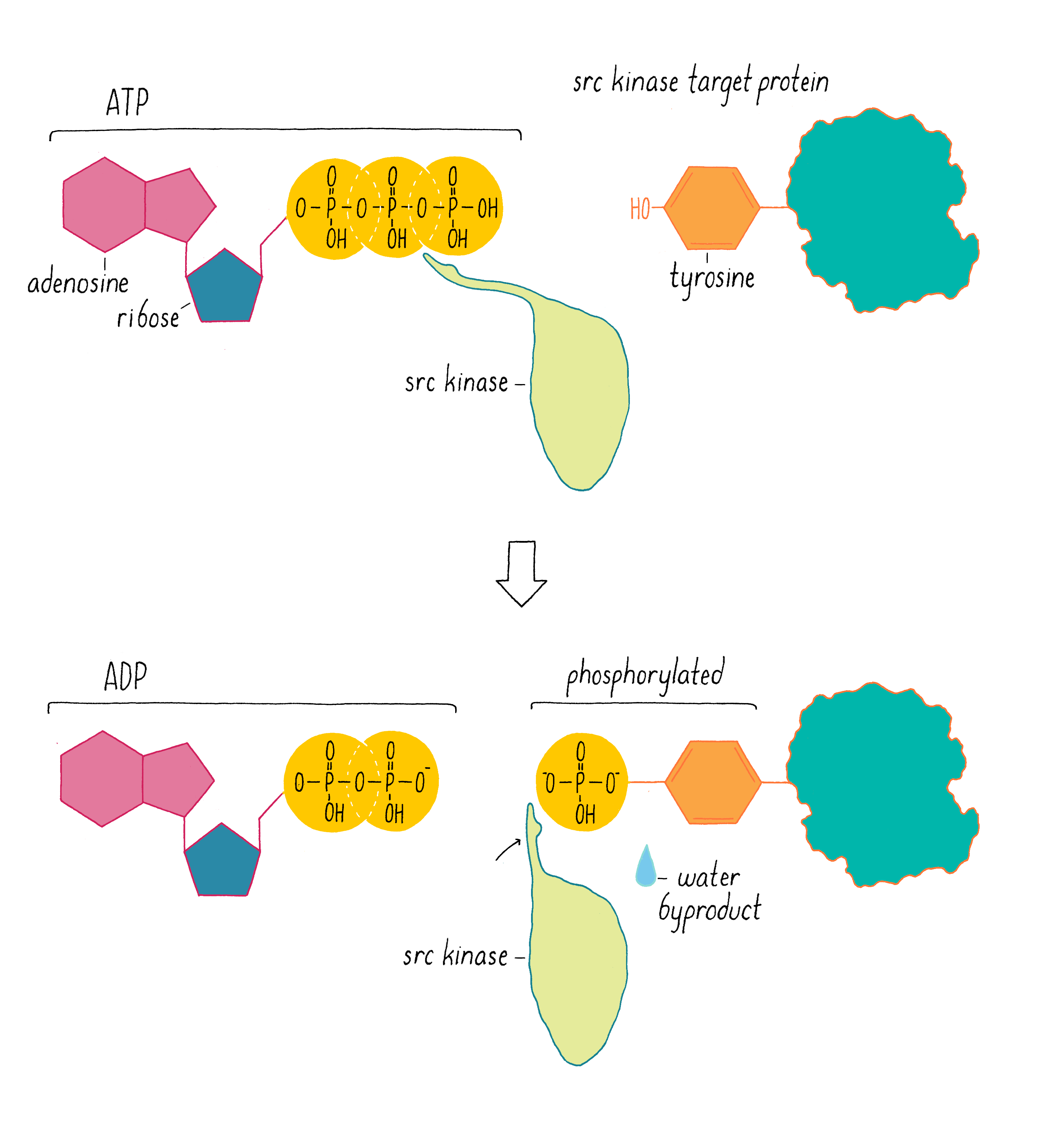
And what about the difference between c-src and the pernicious v-src oncogene? In 1981, we learned why viral src is an outlaw. In most cells, the activity of the c-src kinase is turned off or operating at low levels, and cell growth and division pathways are suppressed. However, v-src has a mutation that makes the kinase permanently active – a gain of function that creates an “oncogene” that drives cancer. We call the normal, well-behaved version of the gene a “proto-oncogene,” meaning that it has the potential to become an oncogene through mutation.
The discovery of the c-src proto-oncogene also triggered an intensive search for similar genes. The inventory of retroviral oncogenes mounted steadily, and it soon became apparent that each of these viral genes had been derived from the genome of a normal cell and made oncogenic through mutation or inordinate expression. Thus, the src paradigm of a retroviral oncogene deriving from a cellular proto-oncogene appeared to be the norm rather than an exception.
It was also easy to imagine that proto-oncogenes could be converted to oncogenes by means other than incorporation into a retrovirus and that cells could contain proto-oncogenes that had not found their way into retroviruses. Thus, proto-oncogenes could be the keys of a keyboard on which carcinogens might play. Indeed, further research revealed the existence of several hundred genes that, when mutated or otherwise unleashed, can contribute to cancer. We now know that every patient’s tumor has a distinctive genetic fingerprint, characterized by mutations in genes that change cells from mild-mannered Dr. Jekyll into out-of-control Mr. Hyde.
What we know now about these cancer genes is described in the Knowledge Overview.
Part II: Knowledge Overview —
The origin of cancer
How cancer arises
Cancer is characterized by a complex variety of abnormal cellular behaviors that promote the growth, survival, and metastasis of tumors. Six examples are shown in Figure 14. The great challenge of fundamental cancer research is to explain how this remarkable change in phenotype is implemented.
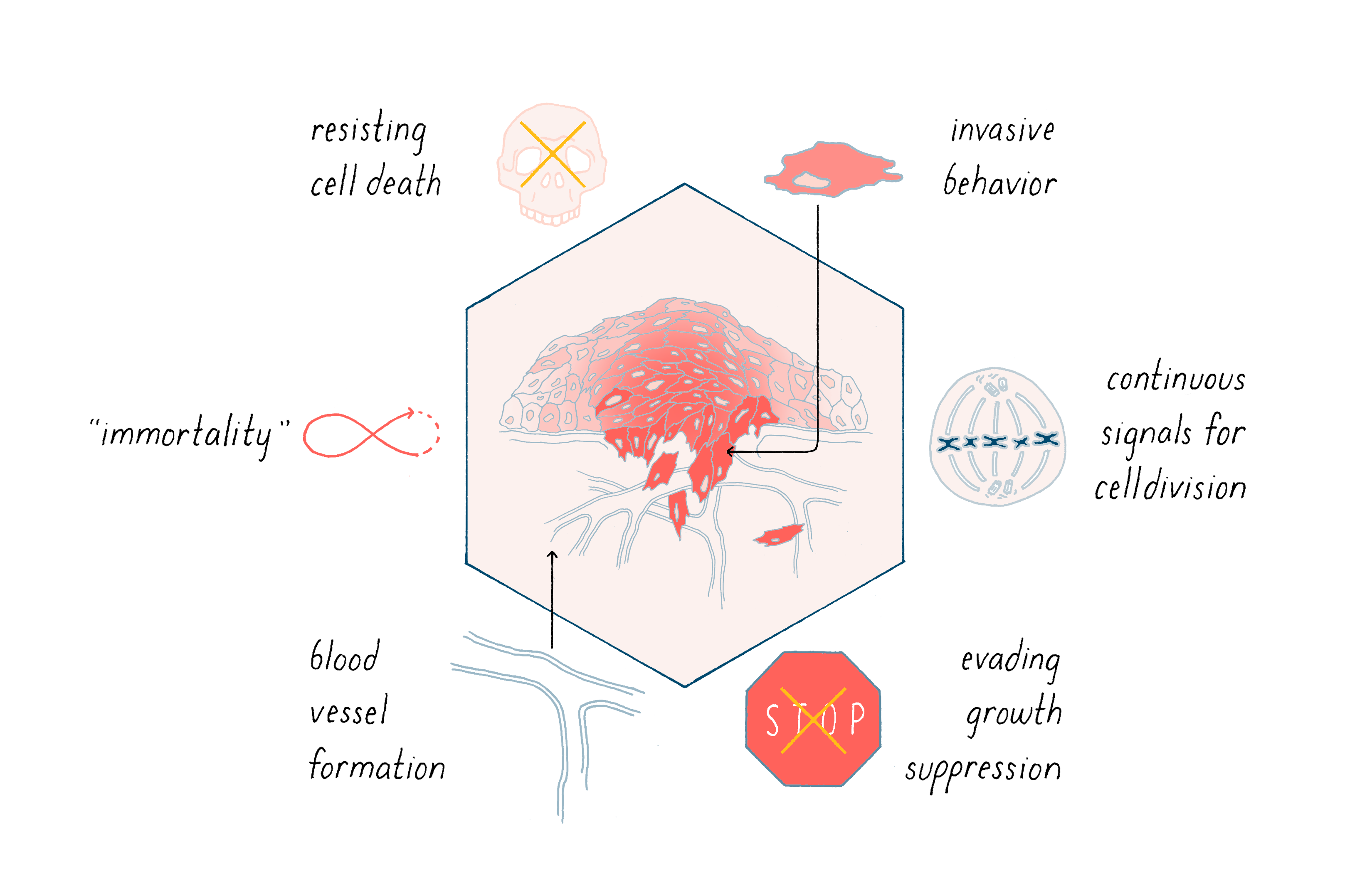
Most of us begin life with well-behaved cells that divide when they should, allowing us to grow into an adult. Cells in our bone marrow also continuously divide to give rise to a constant supply of blood cells, which have a limited lifespan. In all of these cases, cell division is well-controlled. So why do we get cancer?
The reason is that DNA in our cells is constantly subjected to mutations and other disturbances of their activity (see the Narrative on Mutations by Koshland). Mutations can arise through errors in DNA replication, breaks in the double helix, or assaults from the environment (e.g., chemical agents or ultraviolet light for the skin). It is estimated that a typical cell in the human body is estimated to suffer thousands of mutations per day. Most of these mutations fortunately will be repaired by proteins that survey for DNA damage and repair them. However, many errors squeak by. An accumulation of mutations during aging can give rise to cancer. Some individuals have a genetic make-up that puts them at higher risk of developing cancer, like Angelina Jolie. (Heritable predispositions to cancer will be discussed later.)
What do these mutations do? We have come to understand tumorigenesis as a nasty collaboration between two sorts of genetic malfunction. First, gain-of-function mutations turn “proto-oncogenes” into “oncogenes,” which are like “jammed accelerators” (Figure 15). They enhance properties that give rise to cancer, such as uncontrolled cell division and increased invasive migratory behavior in the body. The prototype of this class is c-src, which was the star of the Journey to Discovery. The second class is the tumor suppressor genes (Figure 15). These genes normally function as “brakes” on uncontrolled cell behavior such as division and invasion. In this case, events that cripple or delete these genes inactivate the brakes that keep cells in check.
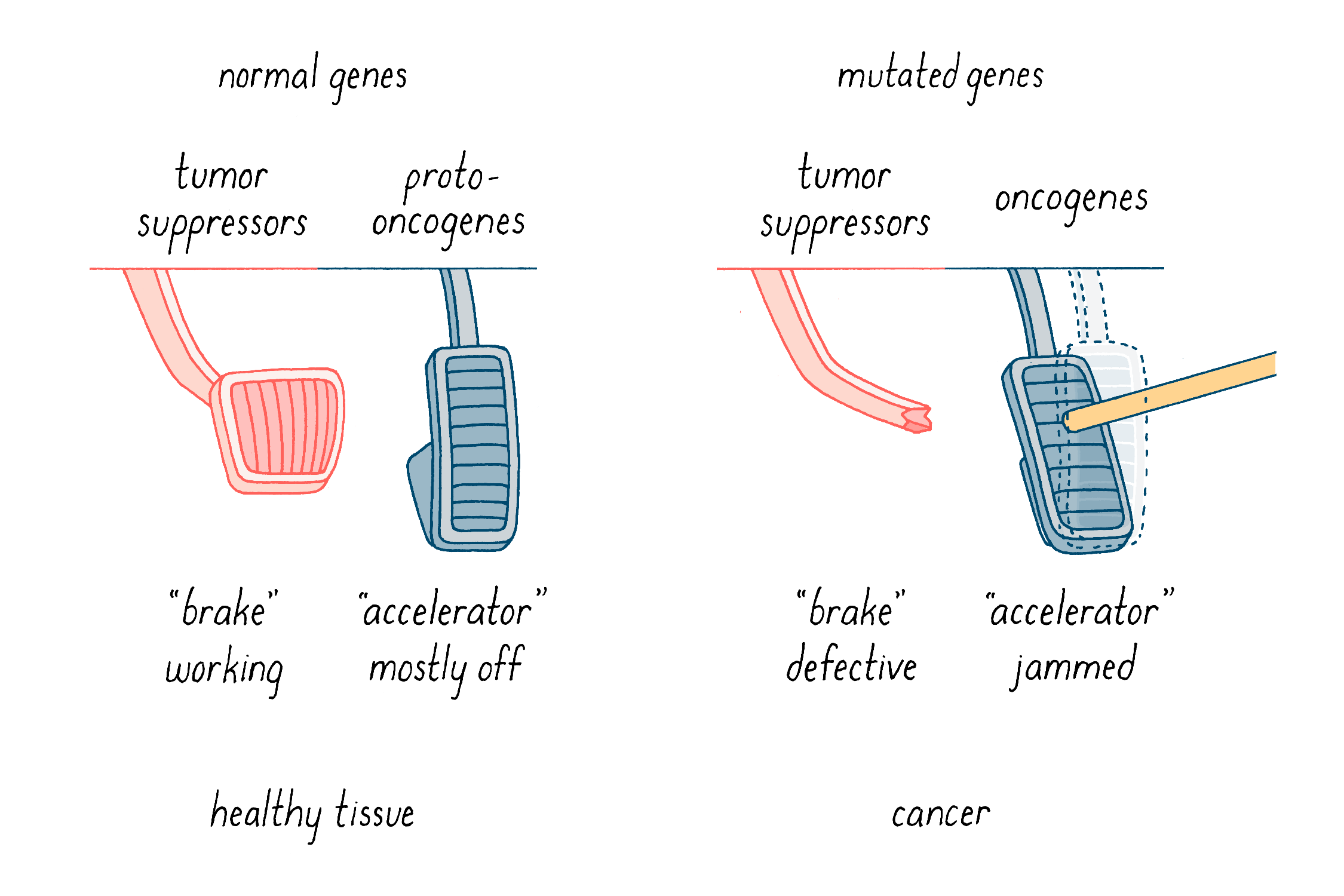
Below, I will discuss how the collaboration of jammed accelerators and defective brakes promotes tumorigenesis, how cancer is thought to arise, and how that knowledge is being applied to our effort to conquer cancer.
Oncogenes: jammed accelerators that drive cancer
There are four well-documented means by which genetic alterations can convert proto-oncogenes to oncogenes, creating drivers of tumorigenesis.
First, and the simplest but nevertheless potent change: a one letter alteration of the genetic alphabet. Such is the case for the ras proto-oncogene. Ras is a “switch” that controls signaling pathways that lead to cell division and other aspects of cellular function. A ras mutation commonly found in tumors changes the twelfth amino acid, which turns the ras switch permanently on. Ras is an accelerator in our scheme, and the mutation jams the accelerator to full speed. Mutations of this sort in ras genes are common in a variety of human tumor types (there are actually three forms of ras in normal human cells, each of which has been implicated in some form of tumorigenesis).
Second, chromosomal translocations (Figure 16) can also create jammed accelerators, but in a different way. An example is the Philadelphia chromosome that I introduced earlier. The translocation responsible for that abnormal chromosome creates a mongrel gene, in which two genes that are normally far from each other on different chromosomes are fused together. The mongrel protein created by this gene fusion acts again like a jammed accelerator that drives the growth of leukemic cells. As you will see in the Frontier section, the jammed accelerator protein created by the Philadelphia chromosome eventually became a target for an extremely successful therapeutic drug.
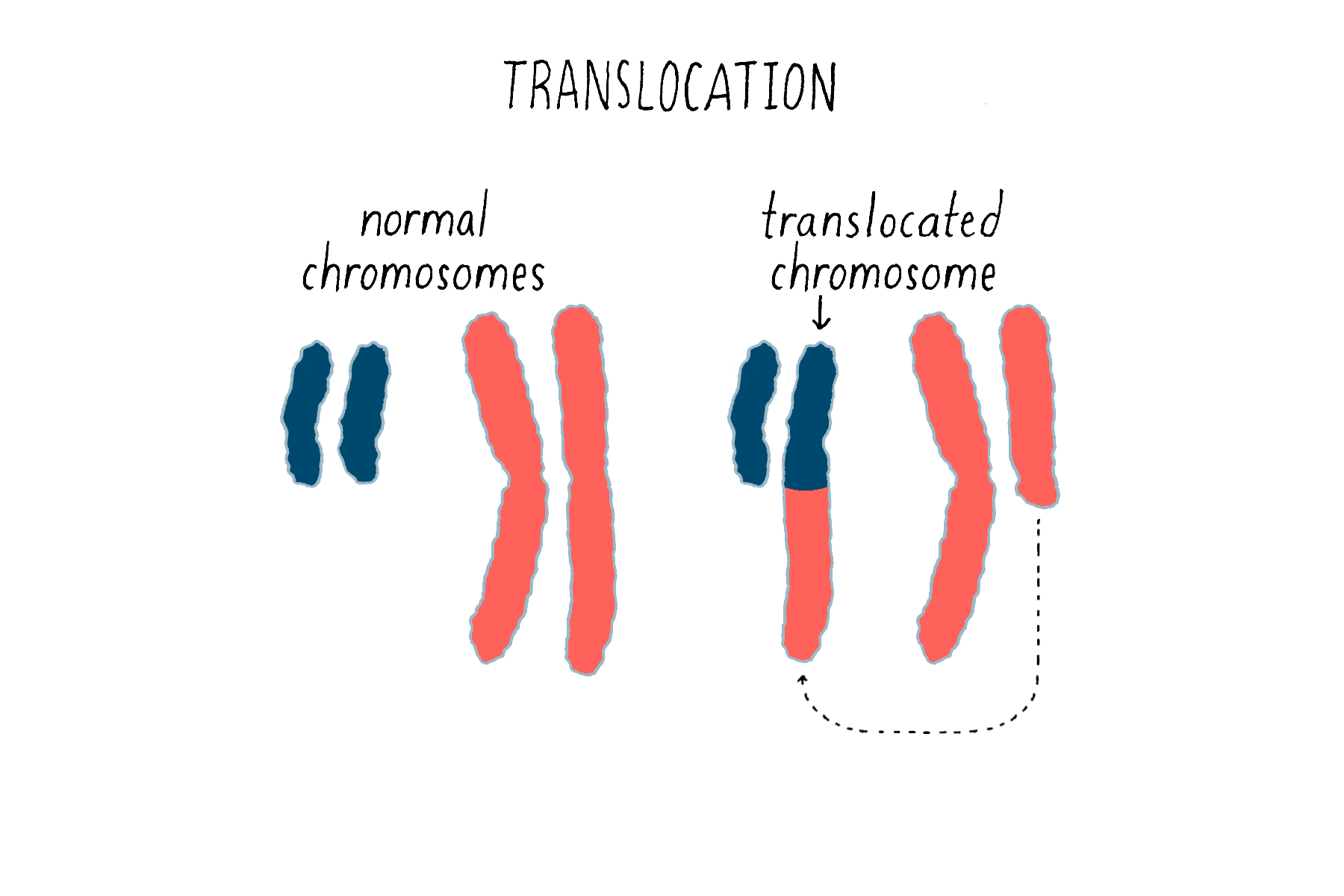
The third means by which genetic accelerators can be jammed involves something known as “gene amplification.” When our cells divide, DNA must be replicated in order to provide a complete set of genes for each of the two progeny cells. On occasion, however, the replicating machinery runs amok, reproducing a region in DNA over and over again – we do not know exactly why this happens. The result can be a huge expansion of a particular region of a chromosome and an extravagant production of the proteins encoded by the affected genes – in other words, a jammed accelerator produced by the overabundance of protein that tends to promote cell division or other features of the malignant cell. This was first observed for the proto-oncogene myc, originally identified as the precursor for a retroviral oncogene, and since been found for other proto-oncogenes. Anomalous amplification of genes can reach a hundred, and even thousand-fold, potentially creating a vast excess of the gene product.
Fourth, controls of gene expression that lie outside the gene itself can be disturbed in ways that unleash expression of the proto-oncogene, creating a jammed accelerator. Although most recently discovered, these disturbances are proving to be quite common, and some appear to be attractive therapeutic targets. They include disturbances of control elements in the DNA of cells, and in the histone proteins that coat the DNA.
Proto-oncogenes that have been converted into jammed accelerators (oncogenes) through mutations or other genetic disturbances are present in virtually all human tumors. With the advent of advanced genomic technologies, the inventory of culpable proto-oncogenes has now mounted into the hundreds.
Tumor suppressor genes: defective brakes on cell division
As the discovery and characterization of proto-oncogenes proceeded, another sort of cancer gene was also coming into view – the crippled brake. The first sighting came from the study of an inherited form of a rare childhood tumor of the retina known as retinoblastoma. This afflicted family tree (Figure 17) illustrates the frequent occurrence of the tumor in two successive generations.

The underlying malady responsible for the inheritance was unmasked in 1983, with the identification of a small deletion in chromosome 13 of those individuals who had inherited the disease (Figure 18). The deletion tracked with inherited retinoblastoma – any children who received it developed the tumor, whereas those who did not receive the defective chromosome were unaffected. The crucial gene removed by the deletion was eventually isolated and christened RB1. Since its absence is the offending malady, RB1 must be a brake rather than an accelerator – the first such brake to be identified in human cells.
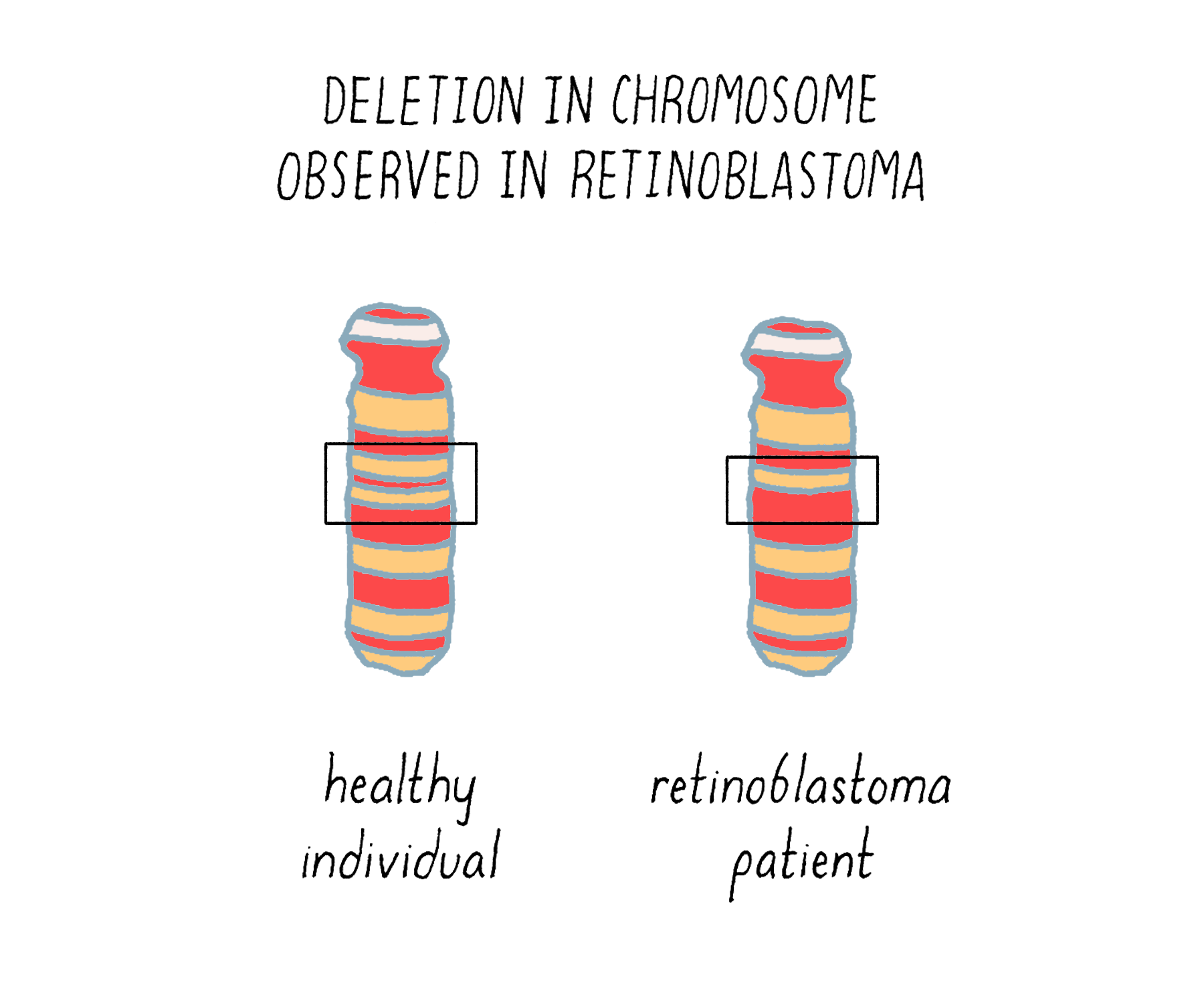
We now understand the nature of the inherited predisposition to retinoblastoma. Children born with a single deficiency in RB1 will have that deficiency in every retinal cell – a head start towards making a cancer. However, the loss of RB1 from one chromosome is not enough to produce a tumor (because the cancer phenotype is recessive). An additional genetic event that incapacitates the remaining normal copy of the gene will launch tumorigenesis (Figure 19). Homozygosity, the inactivation or loss of both copies of the gene, is required for cancer. The likelihood that this will occur in at least one retinal cell is apparently high – the second event is often sufficiently frequent to engender multiple tumors in both eyes. The result is inheritance that has the appearance of genetic dominance. Thus, inherited retinoblastoma is a combination of bad genetics and bad luck.
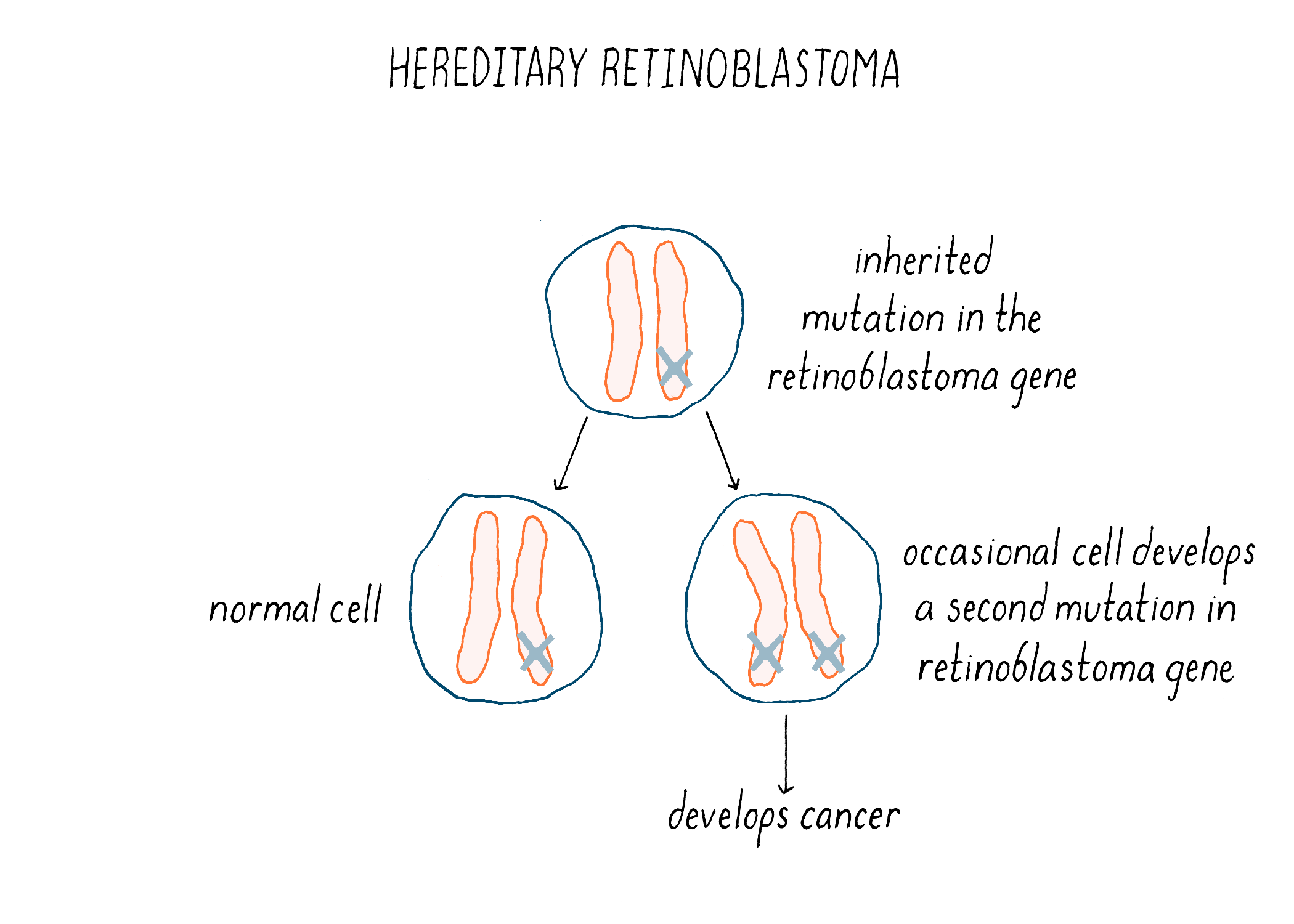
RB1 became the archetype for tumor suppressor genes. Deficiencies in such genes are a ubiquitous feature of human cancer. The inventory of these genes is still growing – their number may resemble that of proto-oncogenes.
Deficiencies in one or another tumor suppressor gene are responsible for virtually all inherited cancer. This brings us back to Ms. Angelina Jolie’s family history of cancer. BRCA1 and 2 are prominent examples of tumor suppressor genes, and a defect in the BRCA1 gene runs in Ms. Jolie’s family. The BRCA genes are essential parts of the machinery that repairs damaged DNA. A deficiency in either of these genes cripples repair and increases the risk of unrepaired mutations occurring in other genes that ultimately produce breast or ovarian cancer. We do not yet understand why these two organs are singled out.
For individuals with a family history of breast or ovarian cancer, a DNA test can be used to assess whether family members have or have not inherited a defective BRCA gene. This enables individuals to understand their cancer risk and decide whether or not to choose removal of these organs, an extreme but effective form of prophylaxis (see the section about Heritable Risk of Cancer below for more on BRCA genes and genetic testing).
Explorer’s Question: Why are heritable cancers usually traced to tumor suppressor genes rather than proto-oncogenes?
Answer:Tumor suppressor genes are recessive by nature. There is no effect unless you lose both of them. Thus, a baby can acquire one defective gene from the parent and develop normally. A later mutation in the “good gene” would drive cancer. In contrast, mutant proto-oncogenes are dominant; even a single mutant allele is generally lethal to embryonic development.
Human tumors caused by viruses
We have seen how direct damage to the cellular genome can give rise for cancer. But there is a large class of carcinogens that often work in other ways – tumor viruses of the sort first discovered by Peyton Rous. Rous made his momentous discovery of a chicken tumor virus in 1909 and openly speculated that viruses might also cause cancer in humans. But it would be 1964 before the first candidate for a human tumor virus was identified, and perhaps not coincidentally, two years later, Rous finally received the Nobel Prize for Physiology or Medicine. Over the ensuing years, more than a dozen kinds of cancers have been linked to human tumor viruses, as shown in Figure 20. We now know that at least 25% of human cancers are caused by viruses, and the number may yet grow larger.

The first example of a human tumor virus emerged from the work of the British surgeon Denis Burkitt, who in the 1950s discovered a previously unknown form of B-cell lymphoma in African children that now bears his name (Burkitt Lymphoma, or BL). Trekking thousands of miles through the African bush to gather case material, Burkitt noticed that BL had a geographical distribution similar to that of yellow fever and malaria. He inferred that BL, like yellow fever, might be a viral disease.
Burkitt was in no position to search for an infectious agent in BL himself. That search was taken up by Anthony Epstein and Yvonne Barr (a graduate student at the time). They scoured specimens of BL shipped to them by Burkitt for signs of a virus or other microbe. They had no luck until they succeeded in propagating the tumor cells in culture (1964). Then, they quickly found an intracellular virus with features of a herpesvirus similar to the type that causes fever blisters. This new virus eventually became known as the Epstein Barr Virus (EBV).
But was EBV the cause of BL, or might it be an innocent passenger in the tumor cells? This question proved difficult to answer for EBV since EBV is widespread in the human population and remains in a latent state for the entire life of most infected individuals. (EBV is also the cause of infectious mononucleosis, a pest of young adults known colloquially as “kissing disease.”) Thus, it was difficult to assemble groups of uninfected individuals for comparison with infected individuals. In due course, however, the data became strong enough to convince most medical scientists that EBV is indeed the cause of BL, enjoying pride of place as the first human tumor virus to be identified. This conclusion is buttressed by several observations beyond epidemiology:
1) EBV induces a B-cell lymphoma when administered to nonhuman primates.
2) Infection with EBV causes cells to divide continuously in laboratory experiments.
3) Activation of latent EBV infection is associated with a variety of B-cell tumors that arise in immunocompromised individuals.
All told, EBV is responsible for at least 200,000 new human malignancies every year – BL and other B-cell tumors, cancer of the nasopharynx, and some stomach cancers. But that number pales in comparison to the havoc wreaked by Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV). Generations of physicians knew HBV only as the unidentified cause of what was then called “serum hepatitis,” attributed to a virus transmitted in transfusion blood and blood products – a disastrous blight on health care. When the responsible virus was finally identified, it became possible to greatly reduce the risk of “serum hepatitis,” now known as Hepatitis B. But the most common form of transmission proved to be close personal contact. Then, alert epidemiologists made the provocative observation that the global geographical distribution of HBV infection resembled that of liver cancer. This led to the supposition that HBV might well cause the disease.
The American epidemiologist (and medical school classmate of mine) Palmer Beasley decisively authenticated the connection between HBV and liver cancer with a study conducted in Taiwan, which had an exceptionally high incidence of both liver cancer and EBV infection, thus facilitating a rigorous study. Beasley and his team followed two cohorts of Taiwanese in what is known as a “prospective study”: one that was chronically infected with HBV (3,454 individuals) and one that was not infected (19,253 individuals). Over the course of several years, there were 61 deaths from liver cancer in the infected cohort, but only 1 death from the same cause in the uninfected cohort. This study provided decisive evidence that HBV can cause liver cancer and is considered a classic in the history of epidemiology.
Any remaining doubt was dispelled with the advent of a vaccine against HBV, which has dramatically reduced the incidence of both EBV infection and liver cancer wherever the vaccine has been effectively deployed. The HBV vaccine, which can rightly be viewed as the first effective cancer vaccine, acts to prevent cancer rather than undertaking the more difficult task of trying to cure it after it has started. But vaccination against HBV alone will never eradicate liver cancer because the more recently discovered Hepatitis C Virus also causes the disease at a devastating rate. Regrettably, there is as yet no vaccine against HCV, so it continues to ravage populations in which careless use of hypodermic needles is common.
The Human Papilloma Virus (HPV) is also a culprit in the genesis of human tumors. The plot for this story began in 1842, when the Italian physician Domenico Rigoni-Stern observed that cancer of the uterine cervix was unusually common among sexually promiscuous women, such as prostitutes, and unusually rare among celibate Catholic nuns. He concluded that the cancer might be a venereal disease, caused by a sexually transmitted infectious agent. At the time, the means to pursue such an agent did not exist, and the idea lay fallow until well into the 20th century. Then, the discovery of EBV and HBV encouraged medical scientists to take up the quest for a viral cause of cervical cancer.
In the face of much initial skepticism, the German scientist Harald Zur Hausen demonstrated that several previously unknown strains of Human Papilloma Virus were the actual culprits – a finding for which he received the 2008 Nobel Prize in Physiology or Medicine. He used molecular hybridization with an HPV DNA probe to detect the virus in tumor cells – the first entry of genomic technology into the hunt for human tumor viruses. In due course, an HPV vaccine was developed, and its widespread use is steadily reducing the incidence of cervical cancer. The vaccine is also given to males, who are the principal source of infection in women and are themselves vulnerable to cancers induced by HPV (throat, anus, and penis). However, there has been a persistent resistance to the HPV vaccine in the United States on the dubious grounds that immunizing adolescents against cervical cancer might encourage promiscuity – the adolescents whom I know laugh at that.
Have all the viral culprits in human cancer been identified? Given that we do not know the cause of many major killers, such as cancers of the breast, prostate, ovary, pancreas, and colon, at least some medical scientists are wagering that there are additional human tumor viruses yet to be exposed. Time will tell. The pursuit is certainly worthy because at the end of each successful chase, there lies the prospect of a vaccine that can potentially eradicate a cancer from humankind.
Tumorigenesis is evolution in miniature
Culprits (oncogenes and tumor suppressor genes) in hand, we turn to the question of how they are deployed during tumorigenesis. Tumors appear to arise in a stepwise manner, orchestrated by Darwinian natural selection in miniature. Two sorts of observations gave rise to this multistep view of cancer.
First, many tumors display discrete morphological stages in their development, progressing from the benign to the malignant in an incremental manner, as illustrated in Figure 21.

Second, the likelihood of most cancers increases over the human lifespan, as illustrated in Figure 22 for colon cancer. It is generally thought that the development of colon cancer requires >20 years from start to finish, which accounts in part for the efficacy of regular screening by colonoscopy. The lengthy time to disease is best explained by the accretion of multiple events.
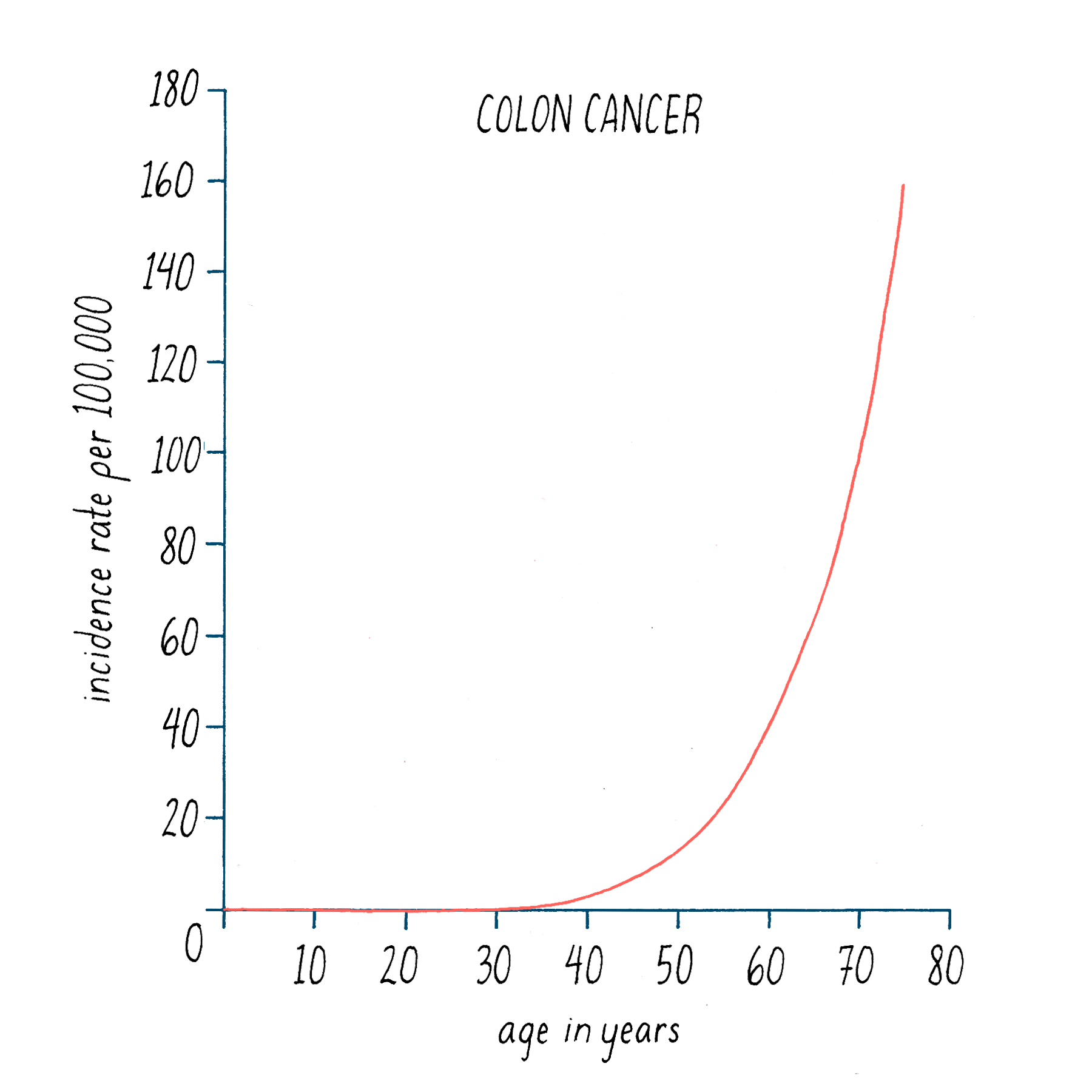
Data of this sort can be used to estimate the number of events that lead to cancer – it is 6 for colon cancer. Overall, the estimates range from 1 in some childhood tumors to a dozen or more in prostate cancer. To a first approximation, the numbers are being born out by the genetic fingerprinting of cancer genomes.
It was Peter Nowell of Philadelphia Chromosome fame who first portrayed the events leading to a full blown tumor as evolution in miniature (Figure 23). In this evolutionary scheme, cancer begins with a single cell that suffers an initial genetic malfunction and proceeds through multiple genetic alterations, each one providing greater survival value to the emerging cancer cell. Each step represents the advent of an additional genetic malfunction. As mutations mount up, independent clones are spawned, diversifying the cellular composition of the developing tumor. The scope of genetic heterogeneity within single tumors can be very large – a recent revelation that raises challenges to taxonomy and therapeutics, which we will explore later. Nowell anticipated this with a prescient comment in the Abstract to his paper: “… each patient’s cancer may require individual specific therapy, and even this may be thwarted by emergence of a genetically variant subline resistant to the treatment.”

The cancer genome at a glance
We have learned that an ailing genome lies at the heart of all cancers. In order to fully exploit this new-found insight, it will be necessary to obtain a complete inventory of the mutant genes in all forms of human cancer, and we will have to sort out the true culprits, the “driver genes.” Driver genes are the ones propelling the misbehavior of cancer cells, as opposed to those that are merely "passengers" – collateral damage from the genetic havoc that takes place during the development of cancer.
At first, the inventory of drivers – proto-oncogenes and tumor suppressor genes – was compiled in a piecemeal fashion. That changed when it became possible to obtain the complete nucleotide sequence of human DNA in a rapid and inexpensive manner. Now, the complete nucleotide sequences of >100,000 cancer genomes are in hand. This is an astonishing achievement that would have been unimaginable when my colleagues and I uncovered the first proto-oncogene, c-src. Although the mining of these data is not complete, we can draw some provisional conclusions about the genomes of human cancers.
First, the cancer genome is typically a wreck – some tumors have as many as 100,000 mutations in their DNA, as well as abnormalities of chromosomal number and structure. On the other hand, there are types of tumors with very few genomic abnormalities, an exception that is not yet understood. Although point mutations in DNA usually come to mind first as the source of driver genes, chromosomal translocations are also present in the vast majority of tumors (see Figure 5 in the Journey to Discovery).
.
Second, the number of driver genes varies from one type of tumor to another, but the range is typically from 5 to 20 – in general, the longer it takes a particular type of cancer to develop, the more numerous the driver genes, although sustained mutagens such as sunlight or cigarette smoke can accelerate the accumulation of multiple drivers and thus produce cancer earlier, melanoma and lung cancer being good examples of this.
Third, there are usually more passengers than drivers, and as I will explain in the Frontiers, it may be possible to exploit passengers in the treatment of cancer, in a manner that would expand and diversify the possible therapies for individual tumors.
Fourth, to date, several hundred potential driver genes have been identified – the emerging cancer cell has a lot to choose from. Indeed, research on laboratory mice indicates that the total repertoire of potential drivers in the mammalian genome could be as large as 2000 – ten percent of all our genes may have some capability in the genesis of cancer, should they malfunction.
And fifth, there is often extraordinary genomic heterogeneity among the cells within a given tumor, presumably reflecting ongoing clonal divergence during the development of the tumor, as illustrated in Figure 23. It is entirely possible that no two cells in a given tumor would have exactly identical genomes. The result is a vast repertoire of distinctive genomes that can harbor a variety of hidden and ominous traits, such as the potential for metastasis and resistance to therapeutics.
The rapidly accumulating knowledge of cancer genomes has enhanced our capabilities on almost every front in the assault on cancer. These include ascertaining the cause and heritable risk of cancer, the classification of tumor types (see Dig Deeper 2), development of new therapeutics, and the prediction of outcome for individual patients. I will survey the state of play for each of these, reserving therapeutics for the Frontiers section because it has so far been the greatest beneficiary of genomic data, resulting in widespread application in the clinic.
The causes and heritable risk cancer
What causes cancer? This is a crucial question. In order to prevent cancers, we must know their cause, and prevention is the most likely way that we might ultimately conquer this disease – “an ounce of prevention is worth a pound of cure.” Regrettably, the determination of cause stands as one of the more difficult forms of cancer research.
Causes have been decisively identified for only a handful of human cancers, and unfortunately, most of the major killers are not among these; for example, we do not know the cause of breast, prostate, colon, ovarian, pancreatic, or brain cancer.
Until recently, identification of cause was largely the province of epidemiologists, who rely mainly on guilt by association. But the advent of genomic tools has opened new avenues of discovery. Genomics has uncovered viruses that cause certain human cancers, as discussed earlier in this section. The genome of cancer cells carries chemical scars that can occasionally speak to the cause of the cancer. For example, skin cancers such as melanoma are littered with chemical damage caused by ultraviolet light, and lesions of this sort are consistently found in driver genes. It has long been believed that extensive exposure to sunlight is the principal cause of melanoma and other skin cancers. Here is powerful evidence for that belief. In other examples, diagnostic chemical scars in DNA reflect the cause of most cancers of the lung (various chemicals in cigarette smoke) and liver (the aflatoxin that contributes to this cancer in certain parts of Asia). In addition, the DNA of tumors caused by chemotherapy bears scars inflicted by the therapeutics.
It is likely, however, that many of the mutations in cancer cells will not be traced to external agents because they are spontaneous in nature – the product of mistakes during the replication of DNA and the failure of surveillance mechanisms to detect and correct the mistakes (see the Narrative on Mutations by Koshland). Remarkably, in some cancers, all of the driver mutations appear to be of this sort.
Heritable risk of cancer
The risk of cancer varies from one individual to another. For example, not everyone who chain smokes for thirty years develops lung cancer. What have we learned about the individual risk of cancer from the study of the genome?
Genetic predispositions to cancer take two forms. One is the inherited defects in certain tumor suppressor genes, which is responsible for perhaps 5–10% of human cancer. One example is the hereditary form of retinoblastoma described earlier in the Knowledge Overview (Figures 17 and 18). A second is the grievous predisposition to breast and ovarian cancer that prompted Angelina Jolie to undergo bilateral mastectomy and subsequent removal of her ovaries (the BRCA1 gene; see the Introduction). There are well established tests for genetic deficiencies in RB1 and the two BRCA genes that can be used in counseling patients and their families (as was done for Ms. Jolie).
There is another form of genetic risk that remains ill defined. Variants of multiple genes can collude to foster an increased or decreased risk of cancer. These genes (or the linked markers that signify their existence) are generally found by the genome-wide association analysis (GWAS) described in the Narrative on the Laws of Inheritance by Tilghman, where the problem of multigene traits/disease is treated in detail. The contribution of each gene alone may be very small, the nature of its contribution to disease is not always evident, and in many instances, the responsible gene has yet to be identified – it is recognized only as a trait linked to a marker in GWAS surveys. Evaluating the risk of cancer imposed by these collusions of multiple genes remains a work in progress. Accordingly, it has not reached patients in any substantive form, and it remains to be seen whether such evaluations will gain general use.
Detection and Prognosis
Early detection
Early detection of cancer is widely thought to increase the success of therapy – next to prevention, it has the potential to be our most potent weapon against cancer. Regrettably, that potential is far from being fully realized. At present, there are screening tests for five major forms of human cancers: breast, cervix, colon, lung, and prostate (see Dig Deeper 3). Of these, only the tests for cervical and colon cancer are generally satisfactory, although the test for colon cancer (colonoscopy) is cumbersome, unpleasant, and expensive. The others are beset by inadequacies and controversies of various sorts. And there are no screening procedures of any sort for major killers such as cancers of the ovary, pancreas, and liver.
Confronted with this relatively bleak record, scientists have turned to genomic analyses as a means for the early detection of cancer. The fundamental strategy is to examine DNA found in body fluids for tell-tale mutations that signal the presence and type of cancer. One approach applies this tactic in an organ-specific manner, testing urine, feces, and perhaps other excretions. In most instances, the efficacy of this "molecular cytology" remains uncertain. But in 2014, the U.S. Food and Drug Administration (commonly known as the FDA) approved a test known as Cologuard that employs analysis of DNA in fecal specimens. Used properly, the efficacy of the test for detection of the various stages of colon cancer appears to be comparable to that of colonoscopy. The next step following a positive result with Cologuard would be colonoscopy to confirm the result, locate the lesion, and obtain a biopsy. But if further experience confirms the reliability of Cologuard, negative results with the test would spare innumerable patients the need for colonoscopy.
Second, there is a more general approach that may eventually allow a single test to detect any latent tumor. We now know that cancer cells and their DNA circulate in the blood quite early during the development of at least some, and perhaps all tumors. Intense efforts are under way to create tests that both detect mutant tumor DNA in circulating blood and indicate the nature of the tumor from which the DNA was derived. Early results suggest that the test can be fortified by adding analysis of blood proteins that might also be signatures of tumors. If adequate sensitivity can be achieved (a big “if” at the moment), this form of molecular cytology could transform the screening for cancer into a simple, inexpensive, one-stop exercise.
Predicting outcomes
Ascertaining the prognosis for individual cancers is vital to the management of cancer patients and can provide some peace of mind for patients. Genome science promises to bring new predictive power to the care of cancer patients.
The first glimpse of this promise came from single cancer genes. For example, in the early 1980s, our research group and that of Fred Alt discovered an amplified gene (given the esoteric name of MYCN) in neuroblastomas, tumors of the nervous system that occur mainly in children. But the gene was amplified in only some neuroblastomas. It soon became apparent that if the gene was not amplified, the tumor responded well to conventional treatment. But if the gene was amplified, the forecast was truly grim, no matter what the treatment (Figure 24). Amplification of the MYCN gene remains one of the most powerful prognostic signs in oncology and is widely used in the care of neuroblastoma patients.

Now it is possible to survey the activity of all 20,000 of our genes in a matter of hours. Using this assay, scientists can search for patterns of gene activity – "signatures" as they are called – that are associated with either good or poor prognosis. For example, surveys of breast cancers uncovered a set of 70 genes whose concerted activity is often linked to poor prognosis. The conclusion is entirely empirical: we do not even know the function of some of the signature genes. A commercial product called the MammaPrint surveys these 70 genes as an adjunct in the care of breast cancer patients. These types of genome-based prognostic tests are far from perfect at the moment but are likely to advance and come into much more common use in the future.
Part III: Frontiers —
Exploiting the cancer genome for therapy
The cancer genome and therapeutics
The genetic paradigm for cancer has completely transformed how we develop new therapeutics for the disease. That story begins with the German medical scientist Paul Ehrlich. Late in the 19th century, Ehrlich developed an antitoxin for the deadly disease diphtheria, an achievement that would eventually earn him the Nobel Prize. Then, he shifted his attention to the use of organic dyes to stain human tissue, allowing him to distinguish among different types of blood cells for the first time.
During the course of this work, Ehrlich noticed that some dyes preferentially stained bacteria as opposed to human cells. This inspired a vision of what he called "magic bullets": agents that could kill infectious microbes while sparing cells of the host. Ehrlich fulfilled this vision by developing a magic bullet for syphilis, an achievement that made him an international celebrity.
But he was not finished. It is rarely noted that Ehrlich's ultimate objective was cancer. Between 1901 and 1905, he tested hundreds of chemicals for the ability to preferentially kill cancer cells. He never found a magic bullet for cancer and died a deeply disillusioned man, telling his wife on his death bed that he had wasted his life – that Nobel Prize notwithstanding.
A century later, we are now in a position to bring Ehrlich's vision to fruition. Cancer genes represent therapeutic targets that are not found in normal cells. We are learning how to attack these targets preferentially and how to annihilate the outlaw cancer cell, while sparing the innocent bystander. There are presently four ways to mount this attack.
First, inhibit jammed accelerators. We know how to do this: development of suitable inhibitors has become a vigorous growth industry. Two examples will be described in detail below.
Second, repair or replace defective brakes. For the moment, this is not feasible, despite the virtuoso techniques for gene editing that are now available. Some inactive proteins encoded by mutant tumor suppressor genes can be revived by pharmaceuticals, but this is an extremely rare circumstance.
Third, use “synthetic lethality” to convert cancer driver genes into lethal weapons – one of the true frontiers in cancer therapy. Work with microbes first uncovered the fact that combining two nonlethal mutant genes in the same cell can sometimes be lethal; the fatal cooperation is known as synthetic lethality. To exploit this phenomenon in the treatment of cancer, drugs are sought that inhibit normal cellular functions that happen to be dispensable under normal circumstances but that kill the cell if it contains one or another driver gene – a synthetic lethal interaction. This approach has been applied successfully to the treatment of breast and ovarian cancers in which a BRCA gene is defective (see Dig Deeper 4 for how this therapy works). Efforts are now underway to find diverse drugs that are not targeted to driver genes but instead are synthetically lethal with them – this effort could appreciably diversify the armamentarium for chemotherapy of cancer.
Fourth, mobilize the immune system to seek and destroy cancer cells. At least some of the mutations in cancer cells cause the cells to appear foreign to the immune system. This could be true of both driver and passenger mutations, thus recruiting the seemingly irrelevant passengers to help eradicate a tumor. Weaponizing the immune system for the purpose of cancer therapy is a rapidly emerging and promising strategy, which has already garnered one Nobel Prize. Immunotherapy of cancer will be covered extensively in another narrative, although you can hear an overview in this whiteboard video (Video 4). But it is not a panacea: resistance to immunotherapy is as common and vexing as that to pharmaceutical drugs.
Targeted therapy of cancer with pharmaceutical agents
Pharmacological inhibition of jammed accelerators produced the first magic bullets targeted at driver genes in cancer. Here, I will explore two examples that illustrate both the success and limitations of this therapeutic strategy: first, the renowned drug Gleevec, a sort of poster child for targeted therapy that has transformed the treatment of chronic myeloid leukemia and several other malignancies, and second, a story about how personalized medicine (in this case DNA analysis of a patient’s tumor) can lead to more successful therapy. Finally, I will conclude with what the modern genomics revolution might offer future cancer chemotherapy.
The development of Gleevec for treatment of chronic myelogenous leukemia
Chronic myelogenous leukemia (CML) accounts for 20% of adult leukemias. Recall in the Journey to Discovery (Clue 3) that David Hungerford and Peter Nowell, in 1961, found that the tumor cells of patients with CML had an abnormal chromosome, which was named the Philadelphia chromosome. Remarkably, nearly all (95%) CML patients had this Philadelphia chromosome, making it a good diagnostic for this particular form of cancer. Janet Rowley then made the important discovery that this abnormal chromosome arises from a reciprocal translocation between chromosomes 9 and 22 (Figure 25).
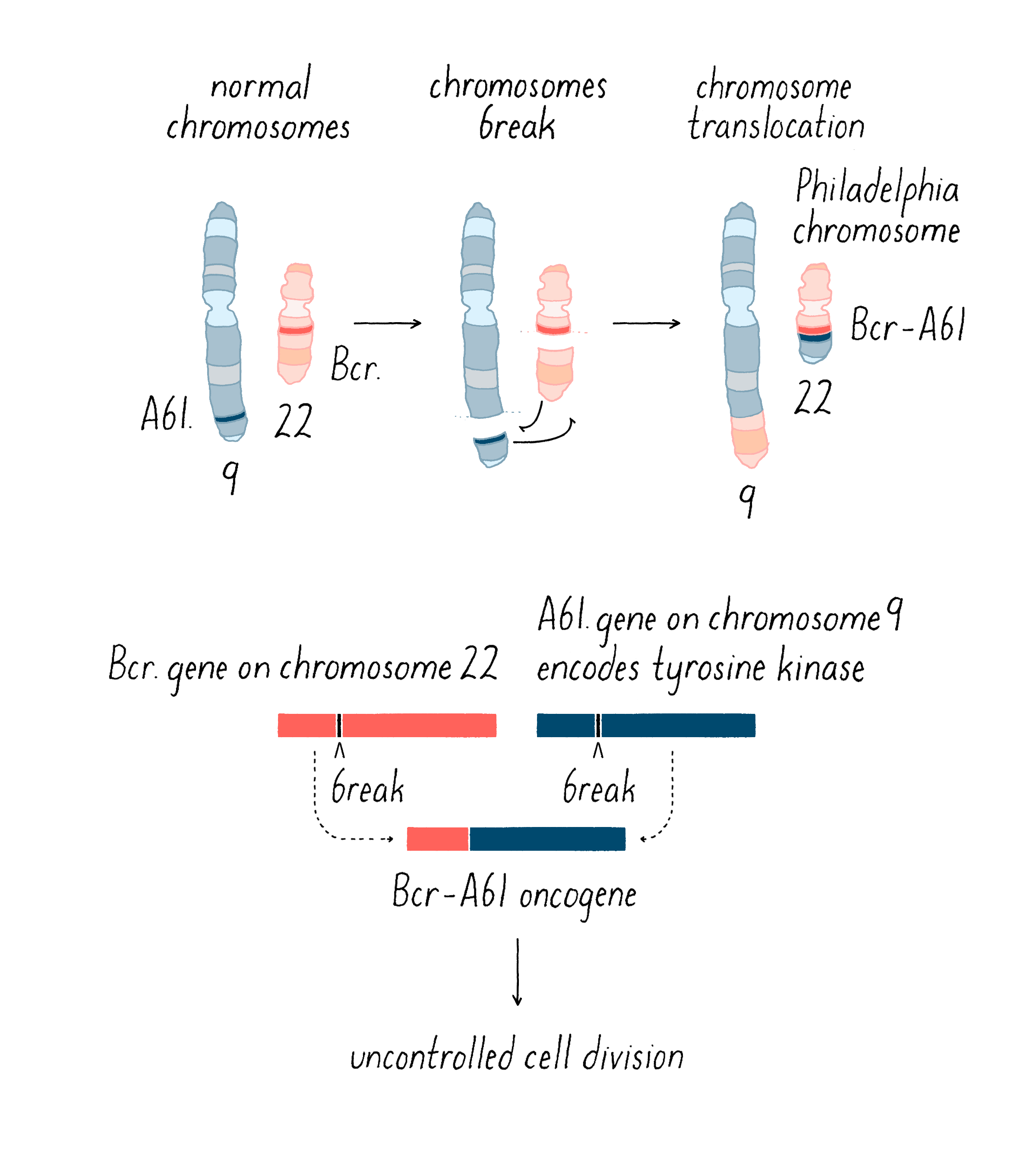
Gene cloning and DNA sequencing brought a new understanding of this disease. The translocation between chromosomes 9 and 22 occurs at specific break points in two genes (BCR and ABL). At the junction, BCR from chromosome 9 is inserted into ABL in chromosome 22 (Figure 25); this fusion event creates a mongrel protein containing pieces of both genes (Figure 25). The Abl portion of the mongrel protein is the interesting actor in this story – it is a protein tyrosine kinase, just like the v-src protein, which was featured in the Journey to Discovery.
When the Abl kinase is turned on, it promotes cell division in bone marrow white blood cells. However, normally, it is turned off, like many other proto-oncogenes. But when the Abl kinase is interrupted by its fusion with Bcr, its normal regulatory mechanism no longer works. Typical of an oncogene product, the translocated Abl becomes “hot-wired” and is always on. The cells now respond to this cue and start dividing.
A blood test of a CML patient reveals the presence of too many white blood cells and that they are not normal white blood cells. Rather, they are an early form (precursor cell) of these circulating white blood cells that normally resides only in the bone marrow. Unlike many cancers, CML does not advance aggressively for ~5 years, which is called the chronic phase of the disease. Later, the CML cells become more abnormal and the cancer takes an aggressive turn, leading to death. We now know that the fusion between Bcr and Abl is the only abnormal gene driving the cancer in the chronic phase of the disease – a “driver” gene as defined above. However, additional mutations occur over time, some hitting additional proto-oncogenes or tumor suppressor genes, and now the cancer takes its more wicked form.
The first treatments for CML involved very toxic and only minimally effective drugs (for example, busulfan, which is still used for other cancers today). The problem is that these cytotoxic drugs are not specific for cancer – they kill normal cells (particularly rapidly dividing cells in the human body, which include the bone marrow, the intestine, and hair follicles). As a result of these toxic effects, conventional “cytotoxic” chemotherapies represent one of the most brutal and dreaded of medical treatments. Less toxic drug treatments were later developed for CML, which modestly extended lifespan but did not cure the disease. Bone marrow transfer from a genetically compatible donor could lead to CML cure. Bone marrow transplants involve ablating one’s immune system in order to introduce a new bone marrow from a matched donor. However, it is dangerous, limited by donor availability, and costly.
Brian Druker, a young physician-scientist from the Oregon Health Science Center, wanted to do better for CML patients. Druker, who studied kinases himself, realized that if one could shut down the problematic oncogene in CML (an out-of-control Abl kinase), then we might be able to stop the uncontrolled division of these cancer cells. Perhaps Druker could find “a magic bullet” that would kill cancer cells but leave normal dividing cells alone, the holy grail of cancer chemotherapy.
It turned out that Ciby-Geigy, a Swiss pharmaceutical company that later became Novartis, had similar thoughts of making a drug for CML by inhibiting Abl kinase. Nick Lydon at the company indeed screened many compounds to find ones that inhibit the kinase activity of Abl. This is a bit like finding a needle in a haystack since one often has to screen tens or hundreds of thousands of chemical compounds to find ones with desired activities, in this case inhibiting Abl kinase activity (see the Whiteboard Video 5 on Drug Development). Lydon found such a compound called STI-571. However, the company seemed uninterested in pursuing STI-57 because CML was too small of a market (compared to other more prevalent cancers, like lung, breast, etc.). We will see later how mother nature thwarted this thinking, at least for the treatment of CML with Gleevec and its relatives.
Druker then teamed up with Lydon and convinced the company to do more tests on STI-571 to determine whether it might be a suitable drug for CML patients. They took cancer cells from CML patients and found that STI-571 inhibited their growth in a cell culture model. They also tested STI-571 in a mouse model for CML and found that it inhibited the leukemia from forming and also did not appear to have bad side effects on the mouse. Furthermore, they wanted to know if STI-571 was specific, meaning that it inhibited Abl but not other tyrosine kinases. They showed that it was highly selective for Abl although it has less potent effects on some other kinases (which turned out to be fortuitous, as described later). STI-571, later named Gleevec (trade name) or imatinib (generic name), works by binding to the same pocket in the protein as ATP, which prevents the kinase from doing its job in phosphorylating protein (Figure 26).

Even with these promising results, the company resisted further development of the drug because of the presumed market size. Druker would not take no for an answer. As an ally, he recruited Charles Sawyers, another young scientist who was also an authority on the mongrel kinase of CML. Together, they successfully lobbied the company to try STI-571 on patients with CML. With the help from the National Cancer Institute (NCI), a Phase 1 Trial was initiated (see Video 5 on Drug Discovery). This stage of a clinical trial is normally planned to test different doses (starting off very low and then escalating) and look carefully for unwanted side effects on patients; it is not intended to measure efficacy or enroll enough patients to establish statistically valid evidence for efficacy. But the Phase I trial results were simply stunning – the cancer just disappeared in the majority of patients. Subsequent larger clinical trials substantiated these results, and the side effects were minor and manageable. In 2001, the U.S. Food and Drug Administration, in response to the overwhelming evidence of efficacy, approved this new drug for use in CML. Gleevec had become a poster-child success story for targeted treatment of cancer.
There are three noteworthy follow-up stories on Gleevec.
The first topic is drug resistance, a topic that is covered in greater depth for AIDS-HIV therapy in the Narrative on Mutations by Koshland. Patients taking Gleevec for years almost inevitably experience leukemia relapse due to the development of clones of tumor cells (see Figure 23 in the Knowledge Overview) that become resistant to Gleevec. Tumors are continuously developing new mutations. Some of those mutations land in the BCR-ABL fused gene, and a subset of those will prevent Gleevec from binding to the kinase, thus allowing the enzymatic activity of the kinase to work normally. How can one get around this problem? The answer is to develop more drugs, which is exactly what the pharmaceutical industry has done, with none other than Charles Sawyers leading the way. Many of these newer drugs will block Bcr-Abl kinase even in cancer cells that have developed resistance to Gleevec.
Second, Gleevec is a multiyear and potentially a lifelong therapy. While Gleevec eliminates all visible cancer from the blood, it appears to not completely cure the disease; a small reserve of cancer cells survives, potentially in the bone marrow. Thus, patients have to remain on Gleevec, or a similar drug, to stay disease free. Returning to the earlier remark that Ciby-Geigy did not think that CML treatment would be profitable, they did not anticipate the possibility of chronic therapy for cancer (like hypertension or diabetes). Early cancer chemotherapy was high-dose and infrequent treatments (sometimes only a few sessions) – not a daily pill for life. Gleevec has proven to be very profitable and raised the possibility that some cancers might be manageable diseases that are amenable to long-term therapy.
Third, being a bit sloppy in its kinase specificity has proven to be advantageous for Gleevec. Another cancer called gastrostromal intestinal tumors (GIST) is driven by activating mutations in another tyrosine kinase called KIT. Gleevec also inhibits KIT and was found to be efficacious in the treatment of GIST.
Gleevec was the first and still perhaps most spectacular proof-of-principle of targeted cancer therapy that makes use of very specific genetic information about the tumor. However, unlike CML, which always shows the same stereotypic molecular defect in the early phase of disease, lung tumors can be very different genetically in different patients; in this case, one may need to understand the genetic drivers for a patient’s particular tumor and then hit it with appropriate “magic bullets.”
Personalized therapies for lung cancer patients
Maki Inada was a talented PhD graduate student at the University of California, San Francisco, where I spent my entire academic career. She graduated and became a professor at Ithaca College. All was well until she developed lung cancer, specifically a non-small cell lung carcinoma. With a 15% survival rate at 5 years, Maki thought that she had received a death sentence. But why her? She was 35 years old, active, healthy, and never smoked. Being proficient at searching Wikipedia and the scientific literature, she read that there were other Asian, nonsmokers with lung cancer who had a particular mutation in the gene for the epidermal growth factor (EGF) receptor. The EGF receptor binds growth factor hormones on the outside of the cell, which then activates an internal part of the receptor, which is … guess what … a tyrosine kinase, just like c-src and abl which we already encountered (Figure 27). Many lung cancers have amplified levels of the EGF receptors that propel the growth of lung cancer cells, but only a small subset of lung cancer patients have Maki’s EGF receptor mutation.
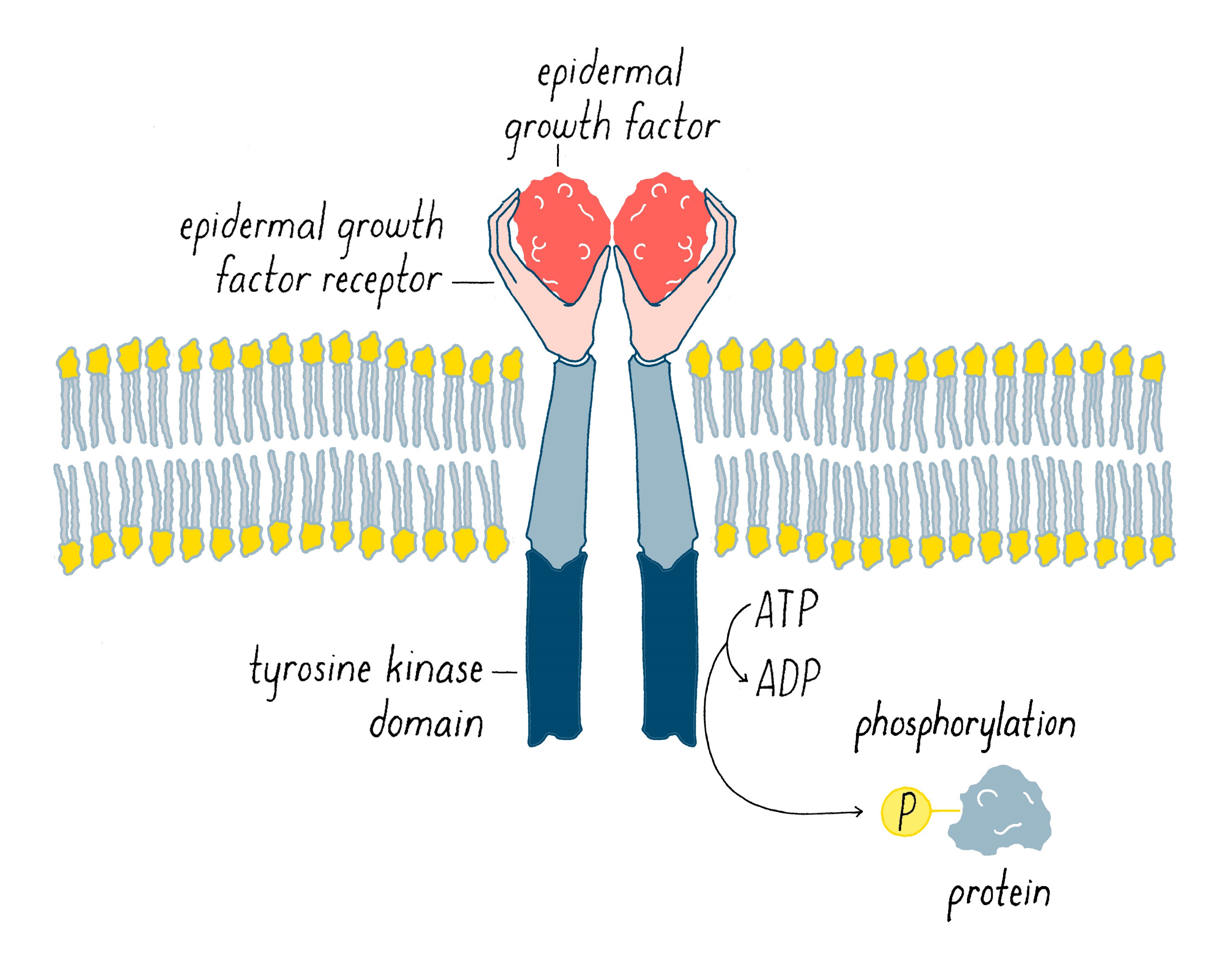
Following the precedent set by Gleevec, pharmaceutical companies began to make inhibitors of the EGF receptor kinase. When inhibitors of this accelerator were first tested in patients, the results appeared discouraging. There was no aggregate prolongation of life. While the overall statistical data were discouraging, however, an occasional patient showed an extraordinary reduction of tumor tissue in the lung during the course of a several week treatment of the EGF receptor inhibitor. Clinical trials were originally conducted with a broad representation of lung cancer patients, 90% of which were smokers. However, those that responded remarkably well were in the 10% nonsmokers. Was there something different about the tumors in these patients? Genome sleuths then entered the picture to try to answer this question. Remarkably, when they sequenced the tumor samples, they discovered that the EGF receptor in the responsive tumors carried mutations that set it apart from the receptor in the unresponsive tumors. Maki’s lung cancer tumor had one of these mutations.
Although it was still in the early days for this correlation between mutation and tumor responsiveness, Maki travelled to the Dana Farber Cancer Institute in Boston to see Dr. Pasi Janne, a leading expert in targeted therapies for lung cancer. There, Maki was able to receive treatment with an EGF receptor inhibitor. And what happened? Her tumor shrank dramatically within 6 weeks of treatment (Figure 28) and then proceeded to melt away after further treatment, with few side effects.

Naturally, Maki’s story is anecdotal, which is not the basis of convincing science. However, the conclusion that EGF receptor mutations respond selectively to EGF receptor inhibitors was supported by clinical studies. In the study below (Figure 29), patients were pre-screened for the presence of an EGF receptor mutation (10% of the total patients with non-small cell lung carcinoma) and if they possessed the mutation, then enrolled for a clinical trial with an EGF receptor inhibitor. In contrast to earlier clinical studies, these patients showed a clear 2–3 fold increased survival with an EGF receptor inhibitor compared with conventional chemotherapy. Patients with non-small cell lung cancer are now routinely screened for the mutations that signify susceptibility to the therapy in order to prescribe the appropriate therapy.

While the targeted therapies work extremely well for many patients with the EGFR mutation, similar to the Gleevec story, resistance often arises within a year or two. Luckily, pharmaceutical companies have been developing next generation drugs that now target the specific resistance mutations. In fact, Maki had a recurrence bearing one of the most common resistance mutations. However, she had survived long enough to take advantage of a next generation inhibitor that addresses her resistance mutation. She was treated with that inhibitor, along with radiation therapy to the area of the tumor, and presently has no detectable tumor. The arms race continues.
Using the cancer genome to personalize cancer therapy
The genomic guidance of treatment described for non-small cell lung cancer represents personalized therapy in the making. We can envision a future when the same strategy will be used for all cancer. The attending physicians would obtain genetic fingerprints of the tumor and assays of gene activity, then use these data to identify potential therapeutic targets and appropriate “magic bullets” to hit those targets. The data may also be helpful in designing immunotherapy for the cancer in question, but for the moment, that is largely speculation.
The data might also reveal whether the tumor is equipped with the apparatus to perform any necessary modification to activate the therapeutic, and whether the tumor possesses the potential to develop resistance to the chosen drug or immunotherapy. All of this information would figure in the design of a therapeutic regimen. Even though the physicians will be armed with magic bullets, they will almost certainly have to use them in combination in an effort to circumvent pre-existing drug resistance – as is commonly done with the less precise therapeutics in current use. To date, most targeted drugs have been studied in isolation (so called “monotherapies”), and in general, responses have been relatively short-lived – the example of Gleevec is exceptional, perhaps because its target is the only driver gene in the early stage of the malignancy. Clinical trials with combinations of targeted drugs are underway, along with trials that combine targeted pharmaceuticals with immunotherapy. The outcomes are not yet clear.
It is a sad truth that traditional combination therapies almost always fail to cure metastatic cancer. Whether the advent of magic bullets and immunotherapy will change things for the better remains to be seen. Perhaps the greatest deterrent to success will be the remarkable genomic heterogeneity of advanced tumors described above, the full magnitude of which has only recently become apparent. Heterogeneity of this sort represents a rich reservoir of potential resistance to both pharmacological and immunological therapeutics, looming as a formidable obstacle to finally winning the war on cancer. In truth, we many never win the war with therapeutics alone. Prevention is likely to play a vital role, should victory ever be achieved (Video 6).
Closing Thoughts
In March of 1986, the Nobel Laureate Renatto Dulbecco published what was among the earliest calls for sequencing the human genome, a project that was at the time deemed either utterly impossible, forbiddingly expensive or not worth the trouble – all of which proved ultimately to be incorrect.
Dulbecco used cancer to justify what proved to be a remarkably prescient proposal. Here in part is what he said:
"If we wish to learn more about cancer, we must now concentrate on the cellular genome ... Research on human cancer would receive a major boost from the detailed knowledge of DNA ... In one generation, we have come a long way in our efforts to understand cancer. The next generation can look forward to exciting new tasks that may lead to a completion of our knowledge about cancer, closing one of the most challenging chapters in biological research."
We have not yet closed that chapter, and we are not even certain how long the chapter may be. But we are turning the pages very rapidly. We are riding a wave that started with the work of Peyton Rous – a telling example of how idle curiosity can spawn a scientific revolution. Rous was never particularly illuminating about the inspiration for his experiments. He explained his effort to transplant the chicken tumor with grafts by saying that transplantation of tumors had succeeded in mammals, so why not try it in chickens. He explained his landmark experiments with filtered extracts of the tumors by pointing out that such work had failed with mammals, so why not try it with chickens. He might never have worked with viruses at all if that Plymouth Rock hen had not showed up on his doorstep. Then, his idle curiosity took over.
The first publications from Rous describing infectious transmission of the chicken sarcoma were studies in prosaic prose: bland descriptions of the experimental procedures, no promotional language, and no grand hypothesis – tedious reading even for me. It had been less than a decade since the existence of viruses had been discovered, and Rous avoided use of that term – he was aware that the concept of a virus remained controversial among biologists. He limited himself to the following comment: “The first tendency will be to regard the self-perpetuating agent active in this sarcoma of the fowl as a minute parasitic organism … But an agency of another sort is not out of the question … For the moment, we have not adopted either hypothesis.” That sort of language would not thrill journal editors today, but it is an admirable display of intellectual caution and honesty.
Obscured by his conservatism was the fact that Rous had made a discovery so far ahead of its time that 55 years would pass before he received the Nobel Prize. He certainly deserved that honor. He had fashioned two momentous legacies: first, the eventual recognition that viruses cause human cancer, a possibility that Rous had toyed with but abandoned when he could not extend his discovery beyond chickens; and second, the chain of research that began with his eponymous virus and eventually linked wayward genes to cancer.
Ironically, Rous went to his grave denying the linkage of genes to cancer. He minced no words in voicing his skepticism, in prominent venues such as his Nobel Lecture and the prestigious journal Nature: “A favorite explanation [for tumorigenesis] has been that [carcinogens] cause alterations in the genes of cells of the body, somatic mutations as these are termed. But numerous facts, when taken together, decisively exclude this supposition” [Nobel Lecture, 1966]. “A hypothesis is best known by its fruits. What have been those [fruits] of the somatic mutation hypothesis [for cancer]? It has resulted in no good thing as concerns the cancer problem, but in much that is bad … Most serious of the results of the somatic mutation hypothesis has been the effect on research workers. It acts as a tranquilizer on those who believe in it ….” [Essay in Nature, 1959]. Even icons have feet of clay.
If you want to read a fictionalized portrayal of this complex and remarkable man, consult the novel Arrowsmith, in which the curmudgeonly character Rippleton Holabird is modeled after Rous. But Nobel Laureate Renato Dulbecco was probably more pertinent in his biographical sketch of Rous, whom he called “a medical man who wished to learn about cancer as a disease and a biologist who did not want to follow the beaten track – willing to hunt for new clues in well-designed but slow experiments.” “Slow” is no longer a welcome pace for most contemporary biological scientists.
Looking back over the course of the scientific revolution ignited by Peyton Rous, I am struck by how it was propelled by personal qualities typical of successful scientists. First, an eye for the main chance: if you see an opportunity to open a door to a secret of nature, seize it. Second, judicious disregard for perceived wisdom: do not hesitate to challenge a reigning dogma that has not been properly tested. Third, a willingness to gamble: playing it safe has rarely (if ever) produced a path-breaking insight. Fourth, faith in the universality of nature: be willing to believe that a chicken virus might hold a key to the heart of the cancer cell. Fifth, choice of an experimental system: despite its humble origin, RSV offered unique advantages among the known retroviruses at the time I first encountered it. Sixth, technical innovation: if there is no known way to do what you want to do, invent one. Seventh, persistence: stay with a problem long enough to be certain that you have the correct answer. And eighth, self-confidence: believe in yourself just enough to risk failure. Having read my story, perhaps you the reader could add to this list.
Rous sarcoma virus was almost lost to history. Following its discovery in 1906, it mainly languished in freezers until 1958, when Howard Temin (a graduate student) and Harry Rubin (a postdoctoral fellow) developed a quantitative biological assay that put the virus into the mainstream. RSV has proved to be a fecund friend of cancer research. Consider its offspring:
– Demonstration that viruses can cause cancer
– The discovery of reverse transcriptase
– The first example of a gene that directly causes cancer
– The role of protein kinases in tumorigenesis
– Phosphorylation of tyrosine, a prime player in cell signaling
– Proto-oncogenes, the first glimpse of a genetic keyboard for carcinogens
– And to date, for what it is worth, five Nobel laureates
Peyton Rous started all this with a chicken, a creature not known for glamor – witness this comment from the German film director, Werner Herzog:
“Stupidity is the devil. Look in the eye of a chicken and you will know this. It’s the most horrifying, cannibalistic and nightmarish creature in the world.”
We have no evidence that Rous feared such derision, presumably because he was undergirded by the biologist’s faith in the universality of nature. In the words of Jacque Monod, one of the fathers of molecular biology, “What is true for E. coli [a bacterium] must also be true for elephants.” As illustrated by my story and other chapters in this book, modern biological scientists use a broad spectrum of species as experimental systems – bacteria, yeast, fruit flies, rodents, birds, plants, primates, and many others in nature. This may be the greatest lesson of all in the story that I have told.
Dig Deeper
Dig Deeper 1 : Rous Sarcoma Virus genome
The genome of Rous Sarcoma Virus contains only four genes that encode for six functional proteins (Figure DD1). Three of these play essential roles in viral replication. The gag gene produces structural proteins that form a shell (called a capsid) around the RNA genome and a protease that is essential to the assembly of the virus. The pol gene gives rises to two proteins: reverse transcriptase (pol) involved in replication of the RNA genome and an integrase (in) that inserts the DNA version of the viral genome into the host genome. The env gene gives rise to two proteins that insert into a lipid bilayer that coats the viral particle and mediates penetration of the virus into host cells. The v-src gene, which is not essential for viral replication, is the oncogene featured in the Journey to Discovery.

Dig Deeper 2: Classification of cancers
The classification of cancers into discrete categories is a venerable pursuit that has traditionally depended upon the gross and microscopic anatomy of the tumor, with an assist from clinical parameters. This is not merely an academic exercise: the results can have important bearing on the choice of therapies and the patient’s prospects (the formal term for the latter is “prognosis”). The advent of genomic fingerprinting has revealed previously unsuspected diversity within various categories of human cancers. In an ever-increasing number of instances, what was once thought to be a single type of cancer has proven to comprise subsets, each of which has a distinctive group of driver genes. For example, traditional taxonomy divides breast cancer into three major types. In contrast, genetic fingerprinting gives rise to more numerous subtypes. This newly emerging complexity could have a profound influence on patient care. See, for example, the section on therapeutics and lung cancer in the Frontiers section.
Dig Deeper 3: Screening for Cancer
At present, there are screening tests for five major forms of human cancer.
First, the venerable Pap test for cancer of the cervix, which searches for suspicious cells in vaginal samples. Fairly effective in itself (it has been saving countless lives for several generations), the Pap test now has a collaborator – a test for DNA of the papilloma viruses that cause cervical cancer. It is possible that the test for viral DNA may someday completely replace the Pap text – at the moment, the two tests are typically used in tandem. In regions where these two tests are not routinely available, the simple measure of coating the cervix with vinegar (acetic acid) serves to identify precancerous lesions, an approach widely used in India and Southeast Asia. The early detection of cervical cancer is one of the few success stories in the early detection of cancer, which ironically might ultimately be rendered obsolete by the vaccine against the HPVs that cause cervical cancer (see the section on viruses that cause cancer).
Second, mammography for breast cancer. Once viewed as irreproachable, mammography has now become the subject of vigorous controversy about the age at which testing should be performed, the frequency of false negatives, and the complications posed by false positives. It remains standard practice in the health care of women, but the uncertainties can pose difficult decisions for patients and their physicians.
Third, colonoscopy for colon cancer. The test is effective, but sufficiently cumbersome, unpleasant, and expensive to deter universal use.
Fourth, the PSA test for prostate cancer, which is falling out of favor as a screening device in the United States, due to appreciable rates of misleading results, and the uncertainty that confounds decisions about when therapeutic intervention is necessary. The test remains generally accepted for monitoring patients for recurrence of prostate cancer following treatment.
Fifth, an expensive radiographic scan for lung cancer, which has evoked controversy about its use for screening and is presently recommended only for those individuals with high risk because they have been heavy smokers. In any event, its sensitivity for truly early disease leaves something to be desired. This is a setting in which testing for tumor DNA in the blood stream might represent a major advance.
Dig Deeper 4: Using Synthetic Lethality to Treat Cancers with a Defective BRCA gene
There are multiple molecular machines that repair mutations in DNA, each by a different means. One of these machines repairs DNA by homologous recombination – essentially, it borrows good DNA to replace the bad (labeled repair pathway 1 in Figure DD2). Another of the machines surgically excises the mistake in the DNA. The BRCA1 and 2 genes encode proteins that are vital constituents of the machinery that repairs DNA by homologous recombination (labeled repair pathway 2 in Figure DD2). When both alleles of one of these genes are defective, homologous recombination fails and more mutations survive, thereby increasing the risk of cancer. When the machinery that repairs DNA by excision is inhibited in cancer cells that are already defective in both BRCA genes, DNA repair is severely compromised. Then, the number of mutations rises to an intolerable point, and the cell dies – in essence, insult has been added to injury (see Figure DD2). This fatal combination represents a “synthetic lethal interaction.” A drug that inhibits the machinery for excision has been approved by the FDA for use against advanced ovarian cancer with a BRCA defect, and the results of recent clinical trials are extremely promising. Data for breast cancer are still pending.

Using synthetic lethality to treat cancers with a defective BRCA gene. See legend of Dig Deeper 4 in the Bishop Narrative on Cancer Genetics.
References and Resources
Guided Paper
Stehelin, D., Varmus, H.E., Bishop, J.M., and Vogt, P.K. (1976). DNA related to the transforming virus gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature, 260: 170–173.
This paper demonstrates that the DNA of normal chicken cells contains DNA sequences closely related to the genes of an avian cancer-causing virus. This knowledge reveals the origins of the genomes of such virus and the roles of these genes in cancer. This research led to the Nobel prize being awarded to Bishop and Varmus in 1989.
DownloadReferences
- Hanahan, D. and Weinberg, R.A. (2011). Hallmarks of cancer: the next generation. Cell, 144: 646–674.
- “My Medical Choice.” Angelina Jolie. New York Times, May 14, 2013 https://www.nytimes.com/2013/05/14/opinion/my-medical-choice.html
- The Emperor of All Maladies: a Biography of Cancer by Siddhartha Mukherjee. 2011. Scribner Publishing.
- The Immortal Life of Henrietta Lacks, by Rebecca Skloot (2011). Broadway Books.
- Manchester, K.L. (1995). Theodor Boveri and the Origins of Malignant Tumours. Trends in Cell Biology, 5: 384–387.
- Dulbecco, R. (1976). Francis Peyton Rous. Biogr Mem Natl Acad Sci, 48: 275–306.
- Nowell, P. (1976). The Clonal Evolution of Tumor Cell Populations. Science, 194: 23–28.
Resources
- The Genetics of Cancer: https://www.cancer.gov/about-cancer/causes-prevention/genetics
This is the National Cancer Institute’s page explaining some of the genetics behind cancer, examples of hereditary cancer syndromes, genetic tests for those syndromes, and much more.
- Genetics and Cancer: https://www.cancer.org/cancer/cancer-causes/genetics.html
This is the American Cancer Society’s page covering how genetics are involved in cancer, how genetics can be used to treat cancer, and much more.
- DNA Science – The Genetics of Cancer: https://www.youtube.com/watch?v=gjQh2lzRsjM This video is a documentary (narrated by Jeff Goldblum) that features some of the original researchers that performed the research mentioned in the Narrative and some of the patients that were involved in those first trials as well as explaining how those treatments were established.
- Brian Druker Describes the Story Behind Gleevec Treatment https://www.ibiology.org/human-disease/imatinib-paradigm-targeted-cancer-therapies/#part-1
Brian Druker is the physician-scientist featured in the Frontiers section for his involvement in the development of Gleevec. In this iBiology video, he describes how this influential drug was developed.
- Gleevec Inhibits Cancer-Causing Kinase BCR-ABL: https://www.hhmi.org/biointeractive/gleevec-inhibits-cancer-causing-kinase-bcr-abl
This short lecture/animation demonstrates how the BCR-ABL kinase works and how the drug Gleevec blocks its function.
- Gleevec-Resistant Form of Kinase BCR-ABL: https://www.hhmi.org/biointeractive/gleevec-resistant-form-kinase-bcr-abl
This short lecture/animation demonstrates how Gleevec resistant BCR-ABL kinases develop and function.
- The Eukaryotic Cell Cycle and Cancer: https://www.hhmi.org/biointeractive/eukaryotic-cell-cycle-and-cancer
This “Click and Learn” interactive diagram demonstrates how malfunctions in the eukaryotic cell cycle can lead to cancer.
- Damage to DNA leads to mutation: https://www.hhmi.org/biointeractive/damage-dna-leads-mutation
This narrated animation demonstrates how damage to DNA can lead to mutations which in turn can lead to the development of cancer.
- DNA Sequence Technology Improves Cancer Treatment: https://www.hhmi.org/biointeractive/dna-sequence-technology-improves-cancer-treatment
This video features Dr. Charles Sawyer talking about how sequencing DNA can be used to create/improve cancer treatments.
- Angiogenesis: https://www.hhmi.org/biointeractive/angiogenesis
This narrated animation shows how cancers cells can develop, proliferate, and recruit blood to the site of the tumor.