CRISPR-Cas9
A New Tool for Genome Editing
Jennifer Doudna, Kevin Doxzen and Martin Jinek
 Jennifer Doudna
Jennifer Doudna
Jennifer Doudna is a professor in the Departments of Molecular and Cell Biology and the Chemistry and Chemical Engineering at the University of California, Berkeley. She shared the 2020 Nobel Prize in Chemistry with Emmanuelle Charpentier for "the development of a method for genome editing." For her studies on CRISPR-Cas9, Dr. Doudna has received several awards including the Breakthrough Prize in the Life Sciences, the Japan Prize, and the Canada Gairdner Award. She has been leading efforts to discuss ethical uses of genome editing technologies. Doudna teaches in Bio 1A, an introductory biology class at UC Berkeley.
 Kevin Doxzen
Kevin Doxzen
Kevin Doxzen, a former graduate student with Jennifer Doudna, is a science communications specialist at the Innovative Genomics Institute, which is advancing genome engineering using CRISPR technologies.
 Martin Jinek
Martin Jinek
Martin Jinek, born in Czechoslovakia and a former postdoctoral fellow with Jennifer Doudna, is now an associate professor in the Department of Biochemistry at the University of Zurich. Jinek received the EMBL John Kendrew Young Scientist Award and the Friedrich Miescher Award of the Swiss Society for Molecular and Cellular Biosciences. He is a scholar of the Bert L & N Kuggie Vallee Foundation and an international research scholar of the Howard Hughes Medical Institute.
What’s the Big Deal?
CRISPR-Cas genome editing might be remembered in history as the single most influential technology in life sciences. For 3.8 billion years, evolution has been slowly sculpting the DNA sequences of genomes of all organisms on Earth. Since the 1990s, we have been able to read the DNA sequences of genomes. With CRISPR, humankind now has the power to rewrite the sequences of at least small regions of the genome. Horrific human genetic diseases such as sickle cell anemia could be potentially "erased" from human genome in the future. Where did this unprecedented tool come from? Most of the engineering was accomplished by nature itself, specifically from a type of immune system that bacteria use to defend themselves from viruses. Scientists first had to understand this system, and once they did, they learned to re-purpose it to serve a new role in genome editing. The experiment described here was a key step in the engineering of a new genome editing technology. Amazingly simple in both idea and execution, it is a clear example of how a well-planned experiment can change the world.
Learning Overview
Big Concepts
Scientists have learned to repurpose a molecular machine, which normally protects bacteria against viruses, into a programmable tool that can be used to edit the nucleotide sequence of a genome. The key experiment that sparked the genome editing revolution is a simple but nonobvious test-tube reaction that could be performed in an afternoon.
Bio-Dictionary Terms Used
Bacteriophage (phage), DNA, enzyme, eukaryote, gene, genome, hemoglobin, in vitro, locus, mRNA, nucleotide, plasmid, prokaryote, RNA polymerase, sickle cell anemia, transcription, transformation
Terms and Concepts Explained
Agarose gel electrophoresis, CRISPR, Cas9, CRISPR-Cas9 genome editing, non-coding RNA, single-guide RNA (sgRNA)
Introduction
-
Doudna and Charpentier met and decided to understand CRISPR, a then mysterious system that allows bacteria to become immune to viruses. At this time, they did not envision its use in genome engineering.
What Events Preceded the Experiment?
-
Scientists earlier found repeating DNA sequences in bacteria, which were named CRISPR (clustered regularly interspaced short palindromic repeats). The CRISPR region produced RNAs (called crRNAs) that were involved in recognizing and cutting the DNA of an invading phage.
-
Charpentier discovered that a second RNA, called a trans-activating CRISPR RNA (tracrRNA), was needed to make the crRNAs.
-
Previous work also revealed an intriguing enzyme called Cas9, which was essential for the bacterial immune system to act against phage. Like the tracrRNA, it was shown to be involved in crRNA production. But perhaps it had other roles? Doudna and Charpentier wanted to know more about Cas9.
Setting Up the Experiment
-
The experimental strategy was to purify components of the CRISPR-Cas9 system and see if they could achieve DNA cutting in a test tube. If they could study this reaction outside of the complex environment of the cell, perhaps they could understand more about how it works.
-
They first tried to see if Cas9 and crRNA (the element that recognizes a specific DNA sequence) would cut DNA. This experiment did not work.
-
The scientists then thought that they might be missing something. They repeated the experiment but now added the tracrRNA. This reaction worked – now Cas9 could cut a target DNA.
-
These results showed that Cas9 enzyme and two RNA molecules, a crRNA matching a DNA target and a tracrRNA, worked together to cut a specific sequence in a DNA molecule.
-
The scientists wanted to simplify the system by designing a “single-guide RNA” (sgRNA), thus reducing the complexity of the system. If it worked, they thought that this simplified system could be useful for genome editing.
Doing the Key Experiment
-
Martin then tested the sgRNA and found that Cas9, armed with an sgRNA, could cut DNA in a test tube.
-
Agarose gel electrophoresis was used to analyze whether the target DNA was cut. This technique is explained.
-
Appropriate control experiments were conducted to ensure that the cutting occurred at a specific DNA site.
-
An additional experiment showed that Cas9 can be programmed with different sgRNAs to cut at very precise locations in a gene.
What Happened Next?
-
These test-tube experiments had broad implications. Cutting DNA at specific sequences in the genome allows for changes in the DNA sequence to be made during the cell’s native repair processes.
-
CRISPR-Cas9 has led to a new biotechnology revolution called "genome editing." Genome editing is now used in thousands of labs around the world and has been applied to study numerous organisms.
-
Practical applications in development include correcting genetic diseases, such as sickle cell anemia, and developing new crops, such as modifying corn to produce better starch.
-
Other proteins can be attached to Cas9 while still allowing Cas9 to target a specific gene. Using this technique, Cas9 has been modified to allow scientists to activate or repress specific genes. Similarly, Cas9 has been modified to enable a specific nucleotide base to be chemically changed into a different nucleotide base. Other modifications enable the location of genes to be visualized within the nucleus.
Closing Thoughts
-
The simple experiment to understand how Cas9 cuts DNA has opened a Pandora’s box of new ethical concerns of how scientists and society should use this powerful new genome editing technology.
Guided Paper
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J.A., Charpentier, E. (2012). A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337: 816–821.
This paper describes the adaptation of the bacterial immunity CRISPR-Cas system into a powerful, programmable, and easy to use tool for genome editing of any species. This research led to the Nobel prize being awarded to Doudna and Charpentier in 2020.
DownloadActivity
To better understand how a gel is run and how it can be used to analyze experiments, try running a simulated gel yourself in the Electrophoresis Activity by Douglas.
Introduction
In 2006, I walked into my office one morning, pulled back the blinds, and gazed across the bay towards the Golden Gate Bridge. Turning back towards my computer, I opened my email inbox and saw a message from a fellow UC Berkeley professor, Jill Banfield. Jill travels the globe collecting environmental samples, which she brings back to Berkeley in order to sift through the dirt and water to extract and sequence fragments of DNA. As I sipped my tea and read Jill’s email, I saw the word “CRISPR” for the first time. CRISPR stands for Clustered Regularly Interspaced Short Palindromic Repeats, which is a unique immune system that allows bacteria to detect and destroy the DNA of an invading virus. Yes, just like humans, bacteria can also become infected with a virus, called a bacteriophage (or phage for short). Jill brought CRISPR to my attention because she knew of my interest in non-coding RNA. Unlike most coding strands of RNA, which are translated into protein, non-coding RNAs tend to fold into unique shapes and serve a diverse set of functions without producing any protein. Two years after Jill’s initial email, my lab began to explore the proteins and noncoding RNAs involved in CRISPR immune systems.
Fast forward several more years to the spring of 2011, when I had a fortuitous encounter at a scientific meeting in Puerto Rico with Emmanuelle Charpentier, a professor from Umeå University in Sweden. As we walked through the streets of Old San Juan during a break in the conference, Emmanuelle proposed a collaboration between our two labs. That conversation would lead to an experiment that would change my life and revolutionize genome engineering.
Emmanuelle had just published exciting results describing a previously unknown RNA molecule that was necessary for certain CRISPR systems. We also knew that a single protein called Cas9 was involved as well. But what did it do? That was the puzzle that we were aiming to solve.
I was excited to get back to the lab and start mapping out these initial experiments with Emmanuelle, her PhD student Krzysztof Chylinski and Martin Jinek, a postdoc in my lab. One of those experiments is featured here. Our motivation was to understand the basic biology of CRISPR immunity. Little did we imagine that our experiments would quickly turn into a revolutionary technology for genome editing. Repurposing the prokaryotic immune system for genome editing has helped accelerate basic research and provided new tools for treating human diseases and engineering sustainable agriculture (see the Narrative on CRISPR by Barrangou).
CRISPR, once an obscure word that appeared in my email, has captured the attention and the imagination of scientists and the public alike. While many people have now heard of CRISPR, not many know about the experiment that launched the CRISPR genome editing revolution. This chapter will explain this relatively simple experiment and discuss how this experiment quickly led to an unforeseen technological advance.
What Events Preceded the Experiment?
When my lab began our first experiments to explore CRISPR immune systems, many aspects of CRISPR biology were known. The description here will be brief. Much more information on the CRISPR immune system can be found in the Narrative on CRISPR by Barrangou.
Starting in the late 1980s, scientists began to notice a recurring pattern of unique repeating DNA sequences in a range of different prokaryotes. Within these genomes, researchers noticed that the repeating sequences were often palindromic, meaning that the sequences of DNA that could be read the same way forward and backward (an example is the palindrome "never odd or even"). Interestingly, scientists also observed that in-between these repeated sequences were short segments of DNA that were an exact match to those found in viruses that infect bacteria called bacteriophage (or phage for short). This observation suggested to researchers that somehow, during a phage infection, the bacterium was able to remove a segment of the viral DNA and insert this piece into the bacterium’s own genome. Scientists simply called the repeated sequences "repeats" and the separating phage sequences "spacers" (Figure 1). Collectively, the long stretch of repeats and spacers were called a "CRISPR array." The ability to update and maintain a record of past phage infections allows CRISPR arrays to function as an "adaptive" immune system, meaning that a memory of past infections enables the bacteria to suppress similar attacks in the future.
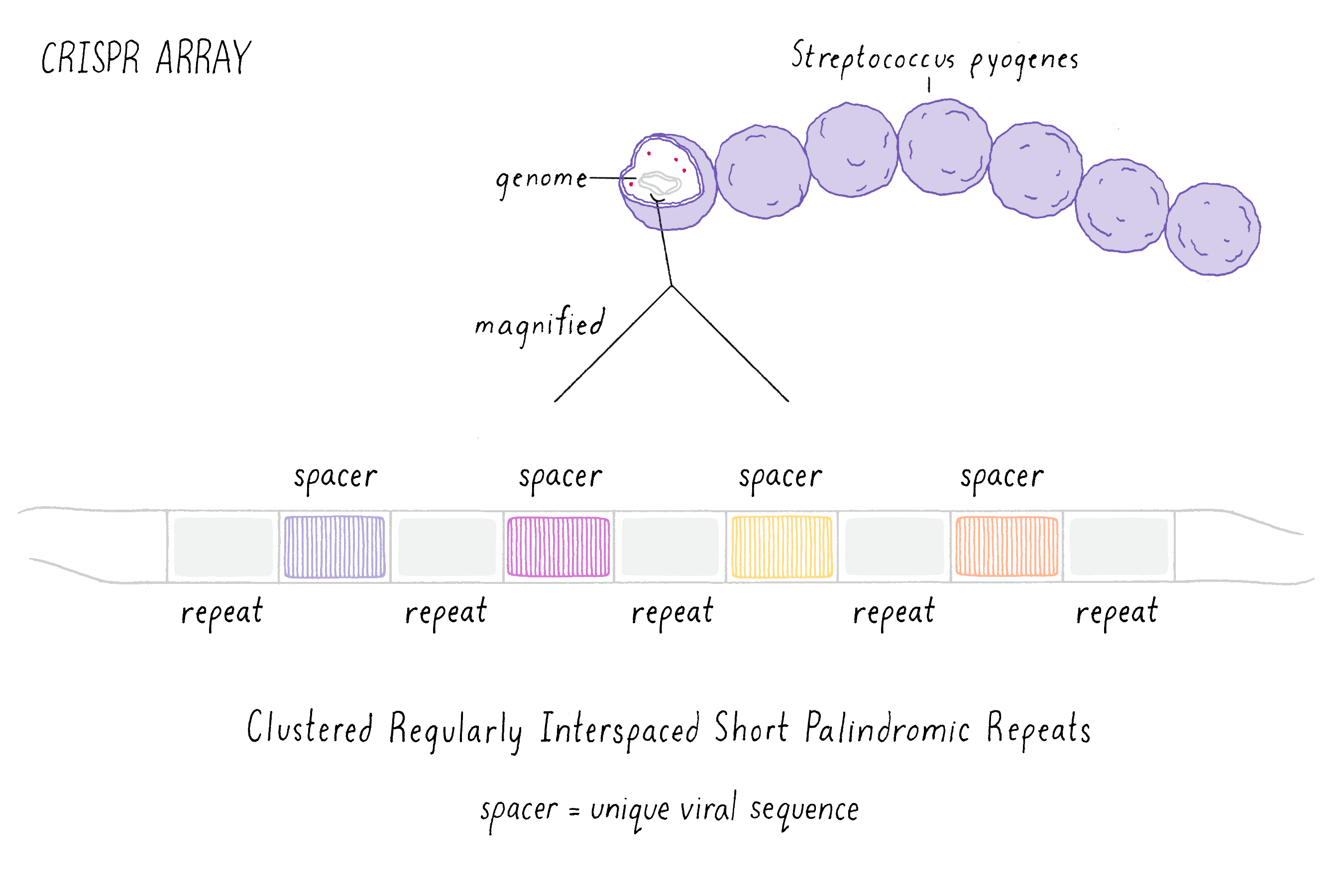
These early observations of bacterial genomes generated more questions than answers. "How do bacteria insert viral sequence into their own genomes?" "How do bacteria use CRISPR arrays as a form of defense?" Scientists thought that protein-coding genes, called CRISPR-associated (cas) genes, found right next to the CRISPR array might hold the answers to these questions (the whole region is called the "CRISPR locus"; Figure 2). There was also good genetic evidence that the Cas protein is essential for immunity against phage (see the Narrative on CRISPR by Barrangou for a broader discussion of CRISPR immunity). But not all bacteria have the same number or sequences of cas genes. This suggested that there are probably several different types of CRISPR immune systems. We decided to pick the simplest system (a so-called type II system) in which there appeared to be just one protein (called Cas9) doing most of the work.
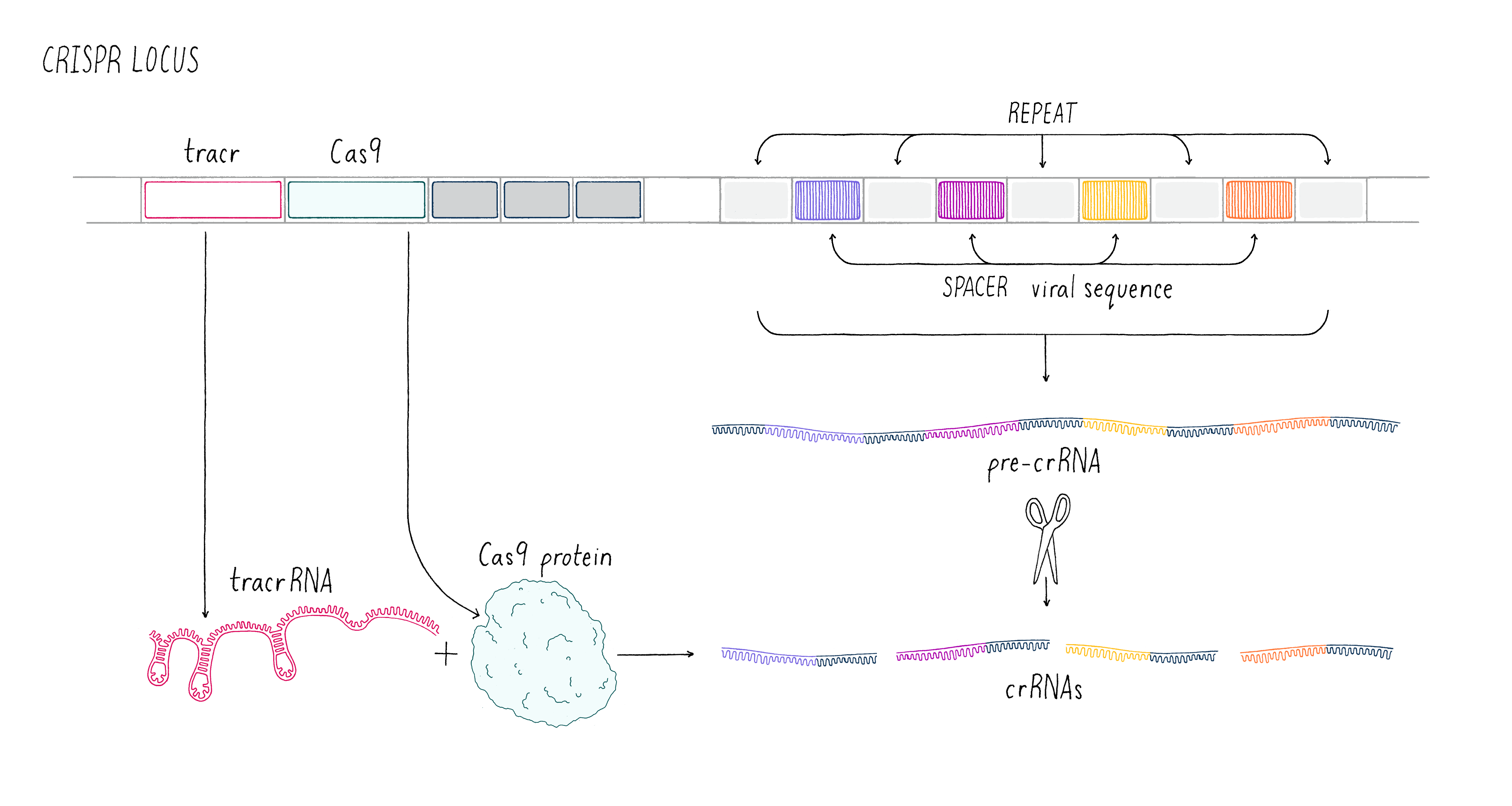
We aimed to understand how the CRISPR array and Cas9 proteins work together as a viral defense system. During a phage infection, the CRISPR array is transcribed into a long RNA that gets processed into smaller RNAs called CRISPR RNAs (crRNAs; Figure 2). These crRNAs contained sequences that match those found in phage DNA, and previous work showed that they are involved in recognizing and cutting the DNA belonging to an invading phage. Emmanuelle and her co-workers revealed that another RNA, called a trans-activating CRISPR RNA (tracrRNA), along with Cas9 and another bacterial protein, was involved in processing the long RNA into the smaller crRNAs.
In summary, we knew of three critical components for viral defense: the enzyme Cas9 and two RNA molecules – tracrRNA and crRNA, the latter which contained specific sequences matching those found in a particular virus. We knew something about how the crRNAs were made. But we still knew little about the next and most important step – how was phage DNA recognized and cut? Was Cas9 involved? As Emmanuelle and I wandered through the streets in Puerto Rico, we were eager to team up and find the answer to this enigma. We had no idea then that the experiments would pave the way for a new idea for genome editing.
Setting Up the Experiment
To answer these questions, our strategy (called "biochemical reconstitution") was to get the entire CRISPR-Cas9 system out of the complex environment of the cell and have it work with purified RNA, DNA, and protein in a test tube. As you will see, the ability to set up various test-tube reactions proved to be a very powerful approach for understanding the CRISPR-Cas9 mechanism.
Making Cas9 protein
For our experiments, we needed to purify the protein Cas9, a task that can be tricky and take weeks or months. Fortunately, an energetic and talented undergraduate student from Germany, Michael Hauer, joined the lab just in time and worked together with Martin to devise the protein purification process. He would only be in Berkeley for four months, but those months would prove to be essential to the success of this project.
Scientists usually use the bacterium Escherichia coli (E. coli) as a factory to produce large quantities of a particular protein. In order to instruct E. coli on what protein to make, scientists introduce a plasmid, a circular piece of DNA, inside the cell. This process of inserting a plasmid into E. coli is called a "transformation." Once inside the cell, the bacteria’s natural machinery reads the plasmid DNA and starts producing large quantities of the desired protein. E. coli can divide roughly every 20 minutes, so after only a few hours, one can obtain billions of cells with millions of protein molecules in each. Next, we break open the cells to release the protein inside. Once this is done, the next step is to purify Cas9 away from other proteins as well as DNA and RNA. Fortunately, our purification worked and Michael and Martin were able to obtain pure Cas9 (see Dig Deeper 1 on how this purification works).
Making RNA
We knew that a crRNA had to be involved in cutting DNA, since its sequence matched that of invading phage (see the Narrative on CRISPR by Barrangou). We would make RNA from a DNA template in a test tube in a process called in vitro transcription. The minimal components required for transcription include a DNA template, RNA polymerase, and ribonucleotide triphosphates (UTP, ATP, GTP, CTP). When all of these components are added to a test tube for several hours, the RNA polymerase will produce millions of copies of a particular RNA (see Video 2).
From failure to success
In our first experiment, we wanted to test if Cas9 was the enzyme that cleaved the phage DNA. As described above, we knew that a crRNA had to be involved. Our first experiment was straightforward – combine Cas9 and crRNA with a target DNA (matching the crRNA) and see if the DNA was cut (we will discuss later the method we used to detect a cut in a DNA molecule). Disappointingly, nothing happened in our test-tube reaction – the DNA was not cut (Figure 3).
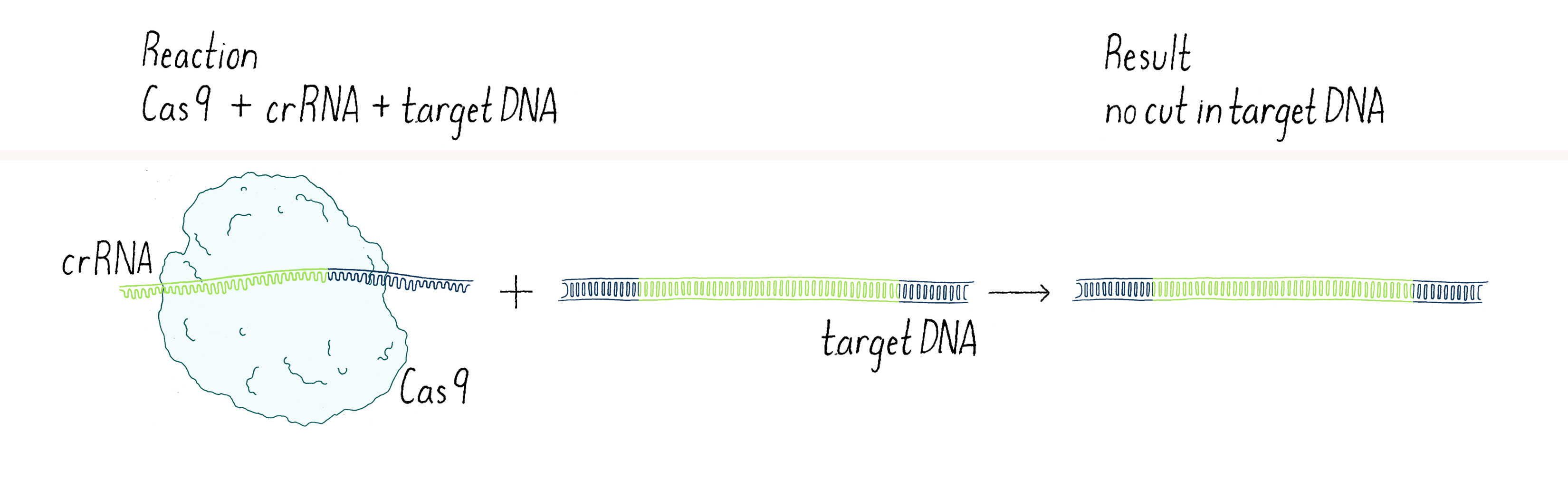
In science, many experiments "fail" (failure is in quotes because you can learn a lot from negative outcomes, as you will see). The key is not to get disappointed, but try to think through what to do next. Why did it not work? Was there a technical problem? Maybe we "killed" the cutting activity of Cas9 when we isolated it (see Dig Deeper 1)? Or perhaps the conditions of our reaction in the test tube were not quite right? Maybe Cas9 was not the enzyme that cleaved the viral DNA? Or maybe we were missing something?
Thinking about the last possibility, our thoughts turned to the mysterious tracrRNA. We knew that it was involved in processing the crRNA. But could it also have a direct role in helping Cas9 to cut phage DNA?
We went back to the lab and set up our reactions again, now with three components – Cas9 enzyme, crRNA, and tracrRNA. To our delight, it worked! With two RNAs (crRNA and tracrRNA), Cas9 now could cut a DNA target. We were now on the right track (Figure 4).
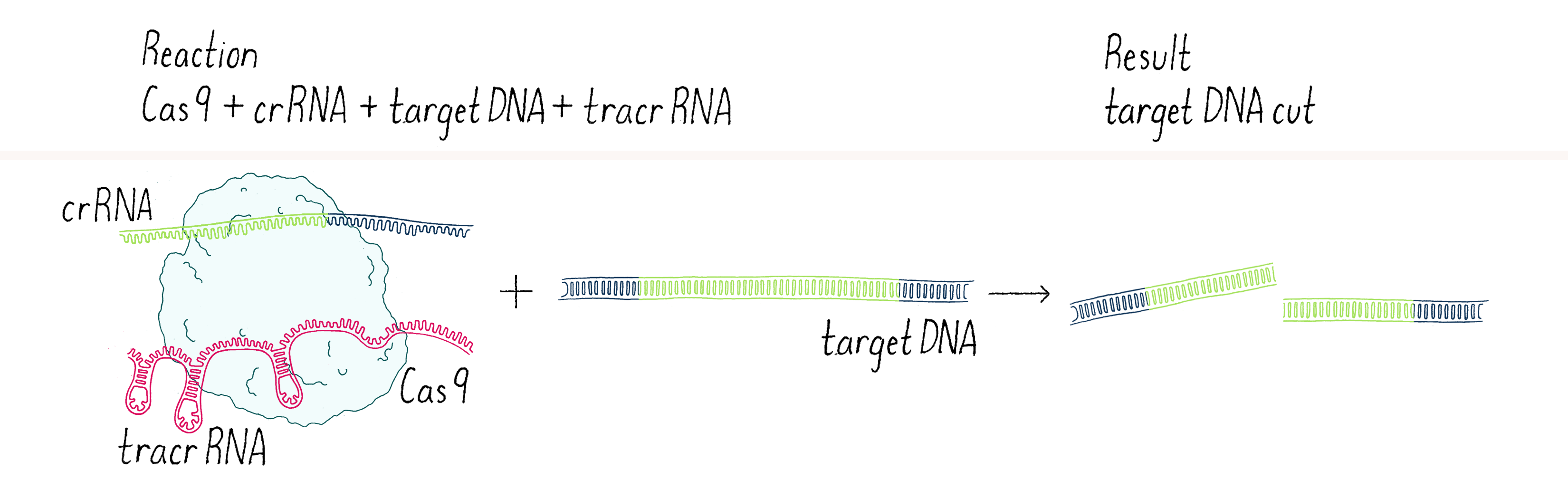
This experiment led us to propose a new model in which three components (Cas9, crRNA, and tracrRNA) come together to form a complex that searches for, identifies, and then cuts both strands of the DNA from an invading phage (Figure 5).
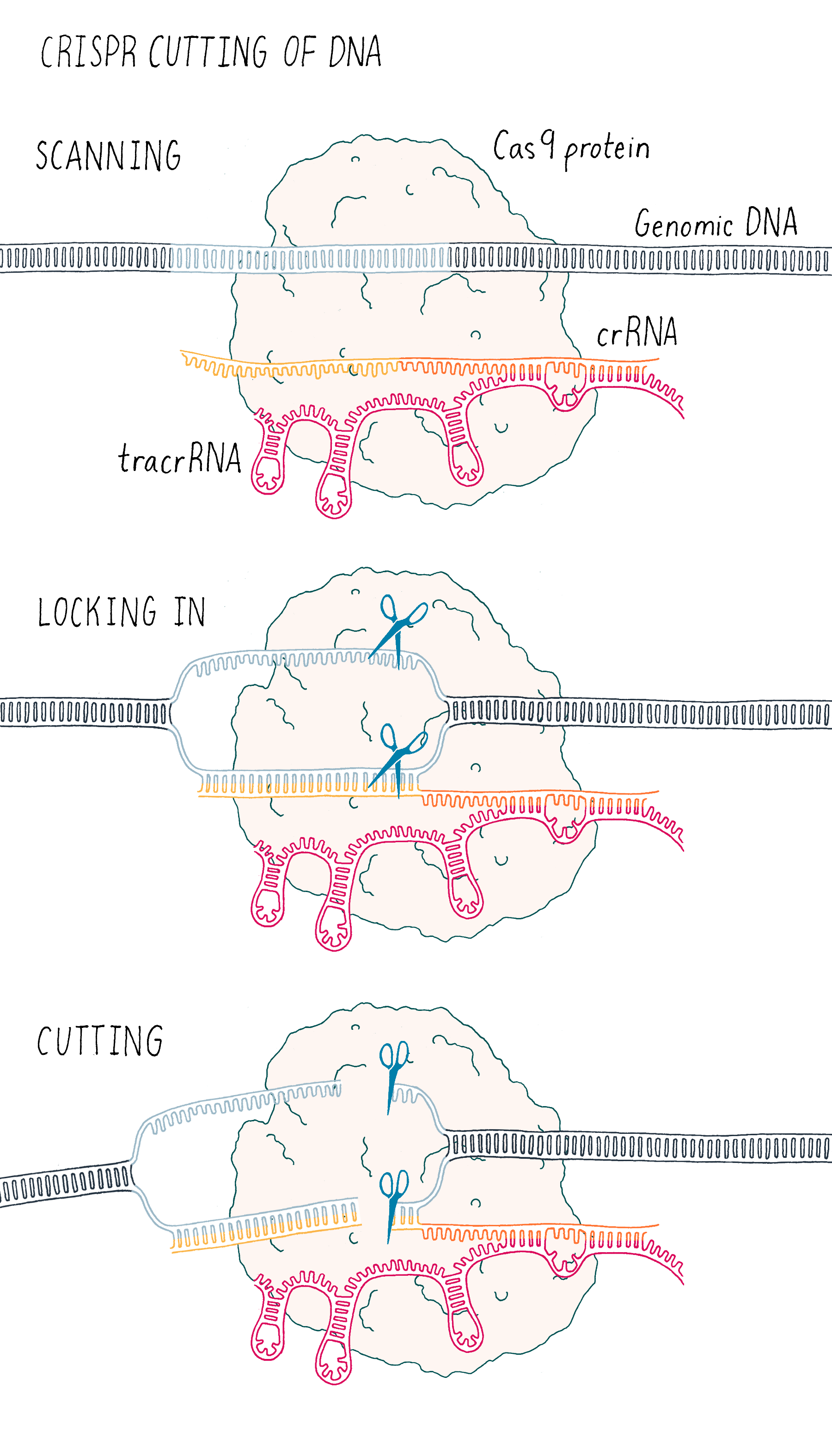
But we were not finished. This exciting result was followed by another eureka moment – could we join the crRNA and tracrRNA into one single RNA that would allow Cas9 to cut DNA? If so, could we then design a simple "single-guide RNA" (sgRNA) to cut any DNA sequence of our choosing?
Why would we want to build such a simple and sequence-specific DNA cutting system? The reason is when a DNA strand is cut, it can be repaired in a way that introduces a new DNA sequence into the repair site. If one could do this in a living organism, then one can potentially "rewrite" the DNA sequence in the genome of that organism. For example, let’s consider a patient who has a single mutated nucleotide in the hemoglobin gene that results in sickle cell anemia. Perhaps one could correct that one nucleotide to produce a normal protein and cure the disease.
In 2011, there were techniques for editing genes, but they were expensive, inefficient, and time-consuming. Thus, creating a simpler sequence-specific DNA cutting enzyme could have powerful applications. However, with three components (Cas9, crRNA, and tracrRNA), this system was not yet as simple as it could be. To make this programmable system even simpler to use, we wanted to reduce the naturally occurring three-component system (Cas9, crRNA, and tracrRNA) down to two components – Cas9 and a single RNA – which would be much easier to express in eukaryotic cells. Here is what happened in the key experiment described in the next section.
Doing the Key Experiment
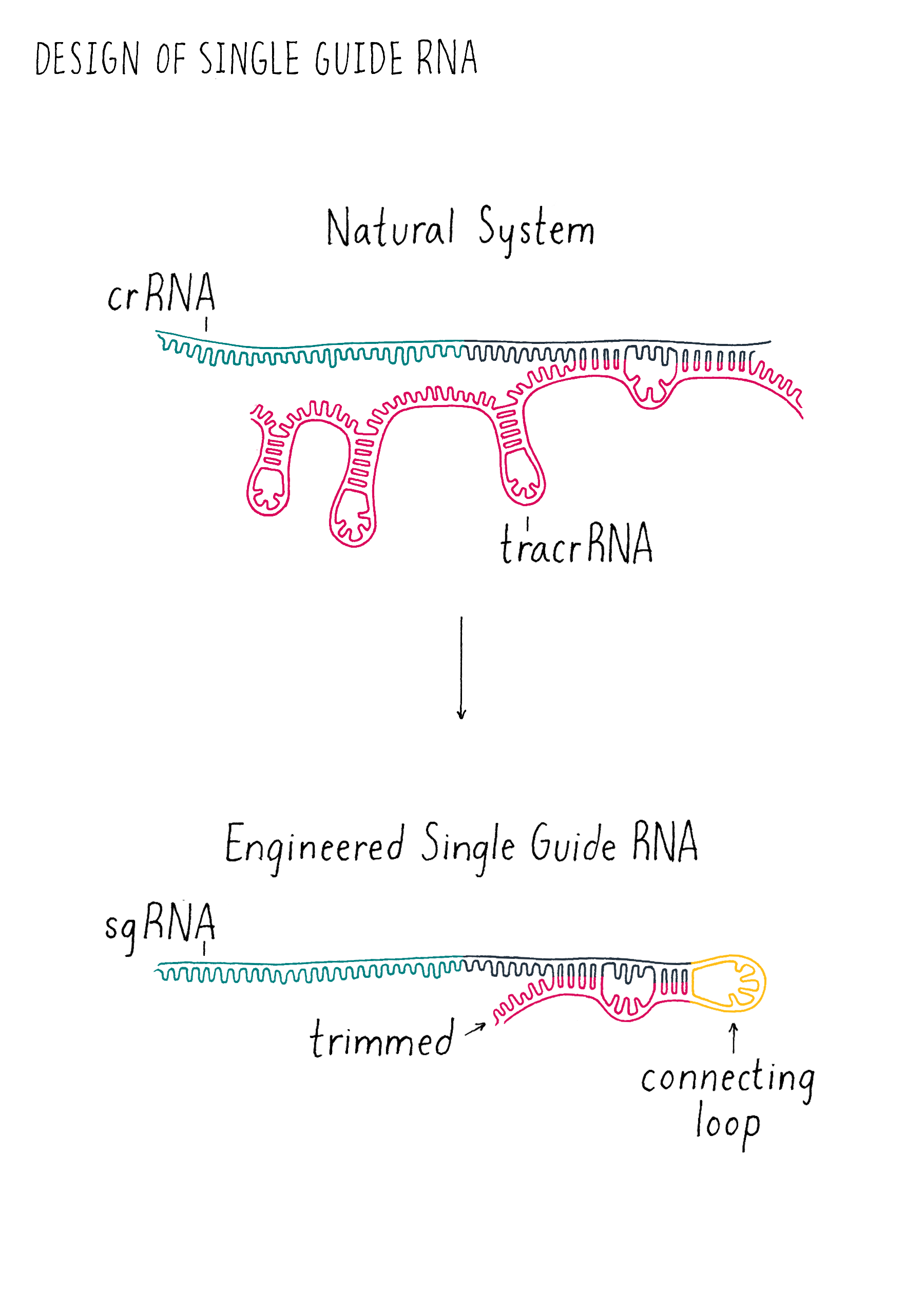
Martin entered my office, and we began to sketch out the sequences of the tracrRNA and crRNA on my white board. When we looked carefully at the sequences, we had a hint that it might not be a totally crazy idea to join them together. One end of the tracrRNA had a complementary sequence to one end of the crRNA. This suggested to us that these two parts might base-pair and form a crRNA-tracrRNA "dual" RNA (essentially the ends of two RNAs would stick together like Velcro).
We decided to add a simple sequence of four nucleotides to link together one end of the crRNA to one end of the tracrRNA to create a "single-guide RNA (sgRNA)" (Figure 6). We also began to draw out how we could trim back the ends of the RNAs that were not linked with the goal of trying to make the system simpler. We decided to test out multiple different lengths of our sgRNA design to see how short we could make the sgRNA and still cut DNA. Since we did not know what might work, we wanted to maximize our chance of success of finding an sgRNA that might operate with Cas9! However, we had to acknowledge potential failure. Perhaps nature evolved the tracrRNA and the crRNA to be separate and that the fusion of the two would not work? We did not know but that is why you do experiments in biology! Even if many ideas don’t work, the few ideas that do work can be transformative, as is the case in this experiment. Let’s let Martin Jinek, who did the experiment, tell us what happened.
Martin
With pure Cas9 protein in hand, Krzysztof Chylinski and I then embarked on a series of experiments to test the DNA cutting activity of Cas9 in a test-tube reaction. Before racing ahead to test the single-guide RNA idea, we first had to establish that our Cas9 enzyme was active with crRNA and tracrRNA and that it was capable of cutting DNA outside of a cell and in a test-tube environment. Fortunately, it was. Now, I could test the experiment of creating a single-guide RNA that was discussed in that memorable meeting in Jennifer’s office.
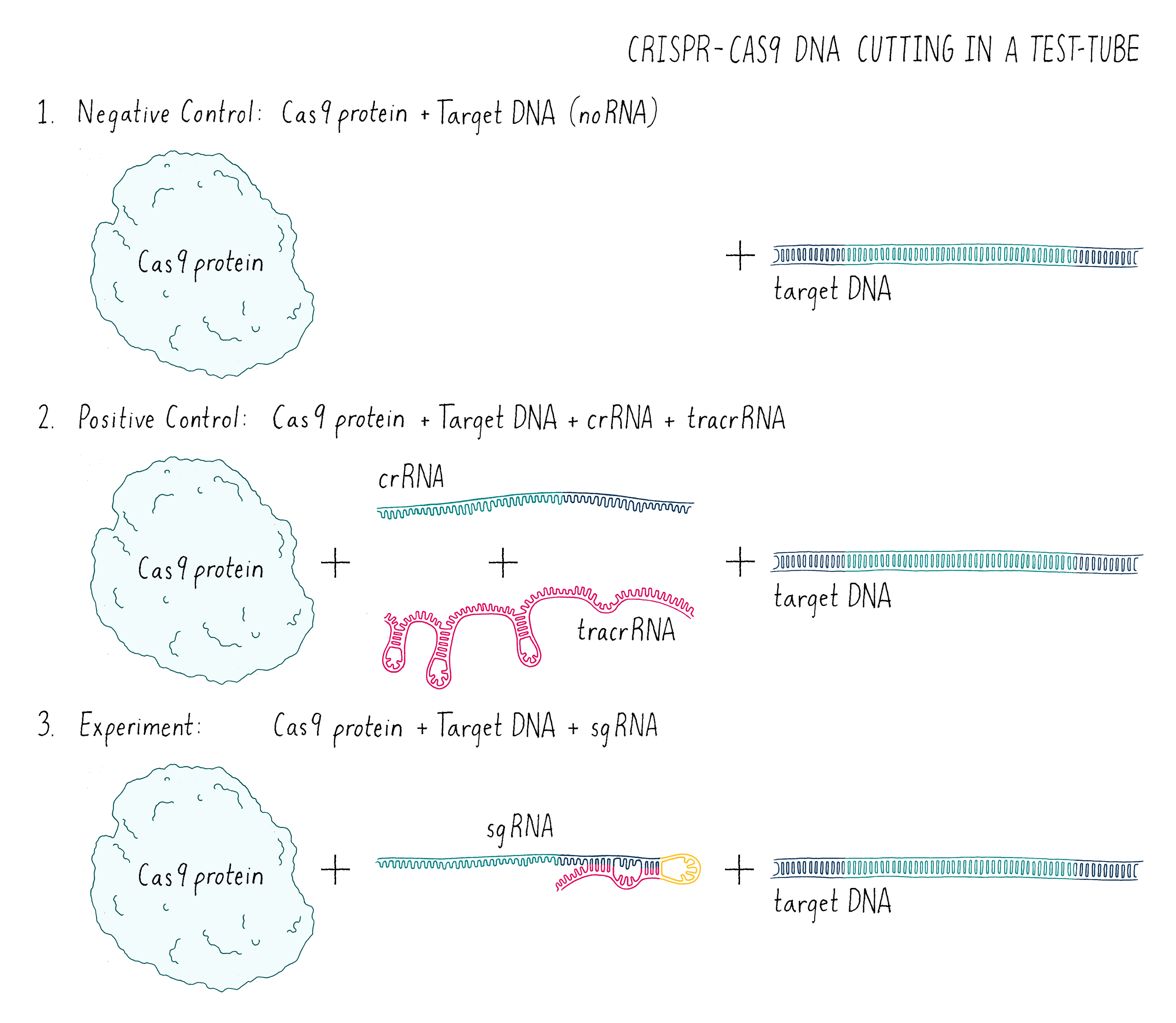
In this experiment (Figure 7), I set up the following reactions:
- A DNA target with Cas9 alone and no added RNAs. This is a “negative control.” The Cas9 should not work without the RNA, so we should see uncut DNA.
- A DNA target with Cas9, tracrRNA, and crRNA. The DNA target had a sequence complementary to the crRNA. This is a “positive control.” From my previous experiments, this combination of factors should cut the target DNA.
- A DNA target with Cas9 and a single-guide RNA that contained complementary segment to the target DNA. This was my experiment. Would it work?
But how can one tell if Cas9 cut the DNA? To answer this question, we used a technique called agarose gel electrophoresis (Figure 8), which can separate two DNA molecules of different sizes. The "gel" is a rectangular slab of agarose that has the consistency of a hard Jell-O. The agarose is placed in a chamber, filled with a salty solution, that has a positive and negative electrode at each end. The DNA is then pipetted into a slot hole (well) in the agarose. An electric current from a power supply is applied, and the negatively charged DNA will migrate through the porous agarose gel towards the positive electrode. A smaller DNA molecule will migrate faster than a larger one because it can maneuver its way more easily through the meshwork of the gel. After an hour or so, the power supply is turned off and a fluorescent DNA binding dye is added to visualize where the DNA migrated in the gel. A smaller DNA will bind less dye and thus will be less bright. Usually, DNA molecules of a specific size move together and will appear as a single band on the gel.
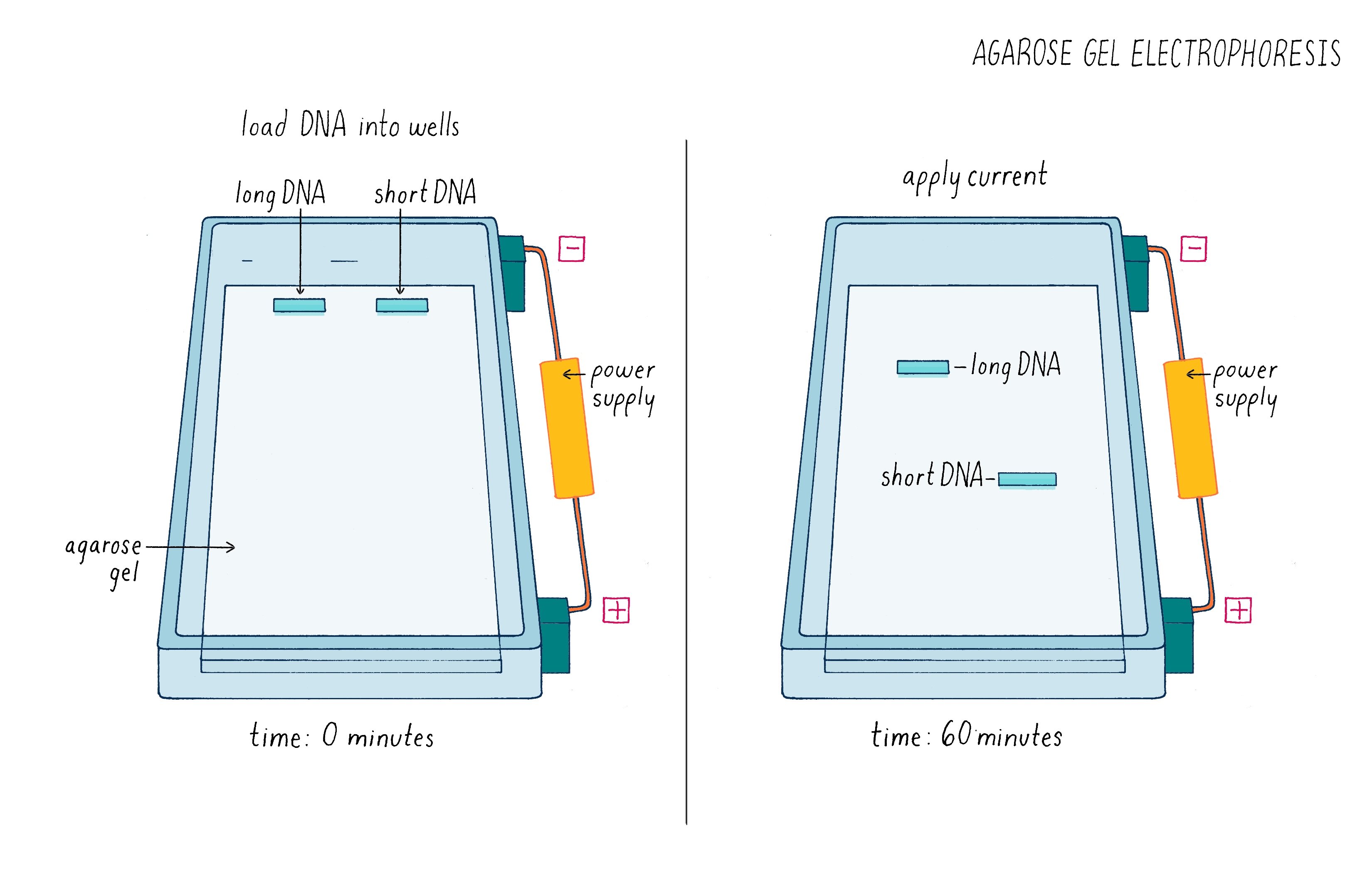
See Video 3 to better understand how a gel is run and how it can be used to analyze experiments such as the one described here. Also run a simulated gel yourself in the Electrophoresis Activity by Douglas.
Now let’s look at the results (Figure 9). The first lane shows a series of DNA molecules of known size. These act as "size standards" by which we can measure the size of an "unknown" DNA molecules produced in our experiment. With Cas9 alone [(1) negative control], the DNA appeared as a single band of the expected size, indicating that that it was not cut. Our positive control (2) also worked – most of the DNA template with Cas9, tracrRNA, and crRNA was cut into 2 pieces whose sizes added up to that of the original target DNA, indicating that it was cut at one site.
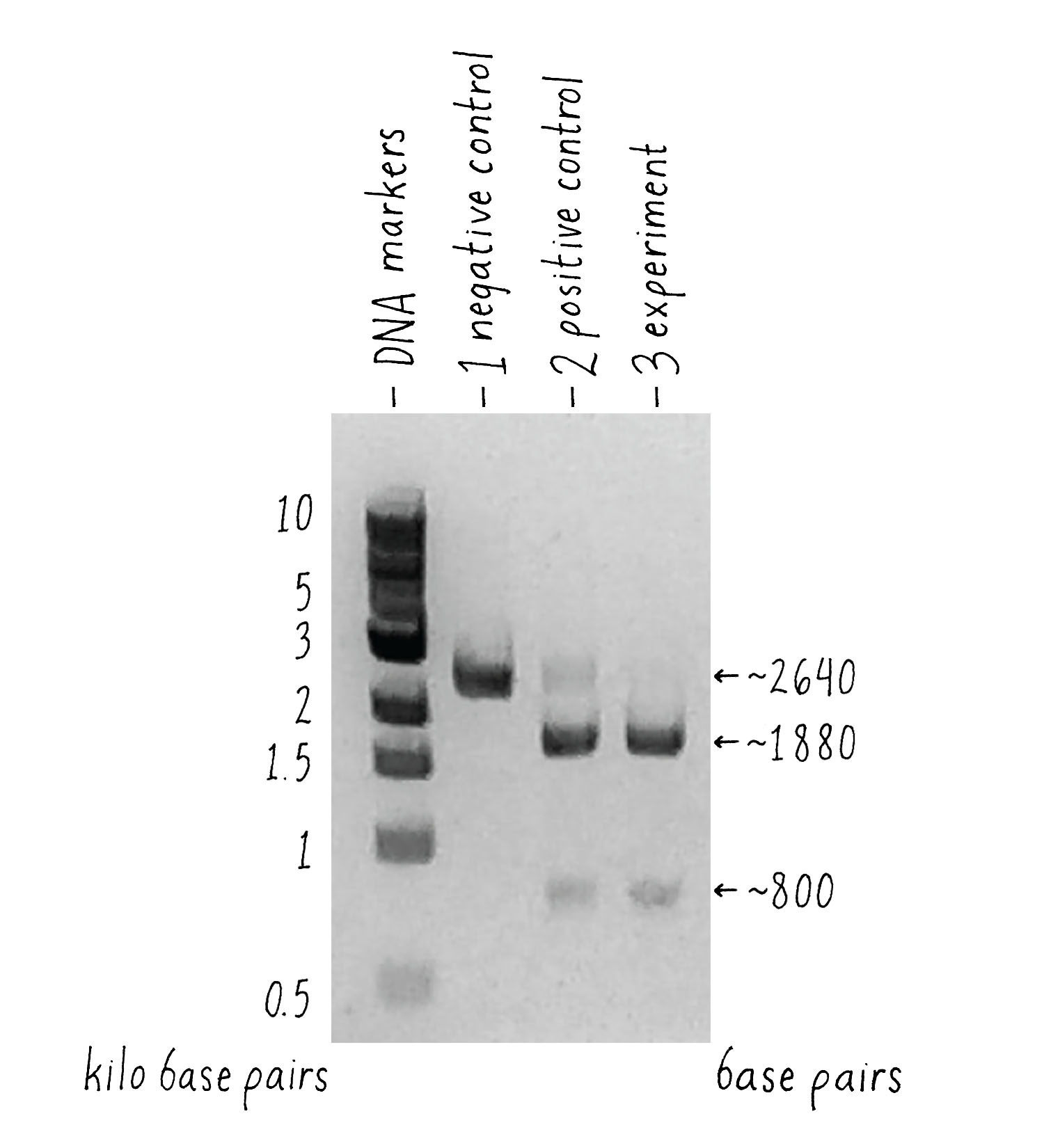
What about the experiment? I was thrilled to see the result! Combining Cas9 with the sgRNA yielded the same cleavage products as the ones that we observed when crRNA and tracrRNA were used. The single-guide RNA strategy that we drew out on the whiteboard worked, and it seemed to be equally or even more efficient than the natural system!
Now it was time for an even more exciting experiment (Figure 10). For Cas9 to become a versatile genome editing tool, we had to test whether the principle was universally applicable. We decided to do one final experiment – design several sgRNAs and test whether they would all be capable of programming Cas9 to cut the correct DNA sequence. To do this, we took the sequence of the gene encoding the green fluorescent protein (GFP) from jellyfish, something that the CRISPR system had never evolved to target. To our great excitement, the experiment worked exactly as predicted on the first try! The designed RNAs were able to guide Cas9 to cut the GFP gene and the sizes of the DNA fragments were as predicted from the locations of the cut sites.
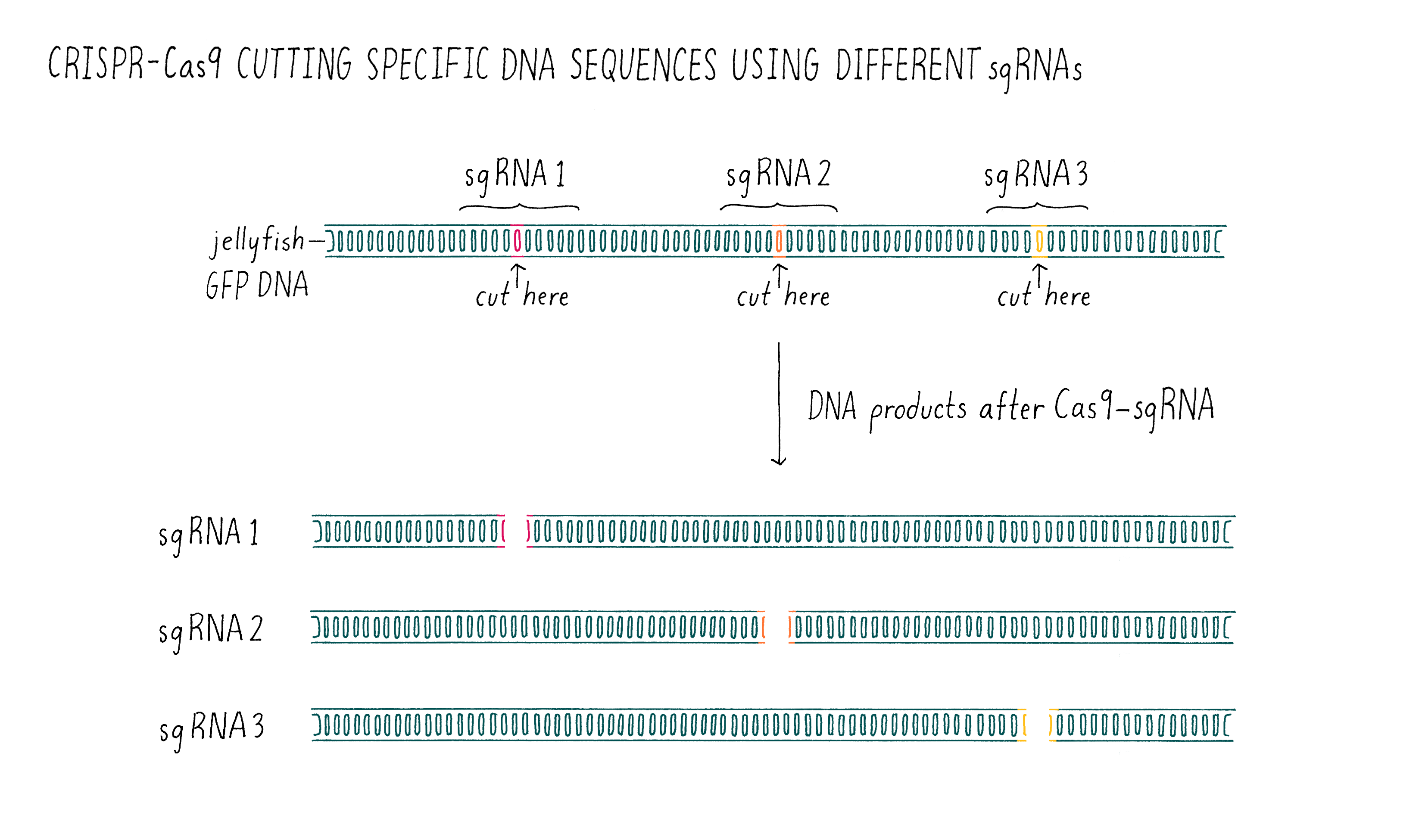
At this point, it was beyond doubt that sgRNAs could be used to direct DNA cutting. It was immediately clear to us that Cas9 is universally programmable, paving the way for its potential use in genome editing of eukaryotic cells!
What Happened Next?
Once we were able to show that we could easily program the Cas9 nuclease to specifically cut any sequence of DNA inside a test tube, we and other labs were eager to see how this tool would work inside a living eukaryotic cell. If Cas9 and a single-guide RNA could target and cut DNA within a human cell, then this method could open the door to edit the human genome and treat human genetic diseases. Many of the over 7,000 human genetic diseases are caused by a single nucleotide mutation within a gene. However, the genome of a human is much larger and more complex than that of a bacterium. In order to find and cut a specific sequence of DNA, the Cas9 needs to navigate through billions of nucleotides to find the correct target. With so many base pairs to check, Cas9 might end up cutting an unintended sequence, called an "off-target." However, following the publication of our "test-tube" experiment, several labs, including ours, showed that the expression of Cas9 and a single-guide RNA would specifically cut DNA in a variety of eukaryotic cells with minimal off-target effects (and scientists are improving the Cas9 enzyme to make the system even more precise).
The ease of using Cas9-sgRNA for genome editing has resulted in this technology sweeping throughout all of biology with unprecedented speed, with many scientists contributing to its development. Before CRISPR technology, scientists could introduce an extra piece of DNA into higher eukaryotic cells with recombinant DNA technology and have it insert randomly in the genome. For example, using a plasmid with its own promoter, Martin Chalfie was able to introduce and express the gene for the green fluorescence protein in worms (see the Key Experiment on GFP by Chalfie). However, scientists could not rewrite the genetic code of higher organisms.
Now with CRISPR-Cas9, the DNA sequence of any gene in the human genome can be modified in ways that can knock out its function completely, introduce or correct a very specific mutation, or add an extra sequence (see the Narrative by Barrangou on CRISPR to understand how this works). For example, in the case of GFP, the DNA sequence for GFP can be appended to the end of an endogenous gene to make the expressed protein fluoresce. This can be advantageous, since the protein will be produced at its natural levels in cells compared to using plasmids, which tend to produce the protein at much higher levels.
The beauty of CRISPR-Cas9 is that it works in a huge variety of species. Many species that have been considered incapable of genetic alteration by scientists can now be genetically manipulated with this new technology.
CRISPR is being used to develop new varieties of plants with desirable traits, such as drought resistance, pest resistance, or other qualities that will increase desirability or agricultural yield. In contrast to previous genetic modifications in plants, CRISPR causes more specific genetic changes and does not need to include the addition of DNA from a different organism. The first CRISPR crop expected to be commercially available is a waxy corn that has increased amylopectin content. Amylopectin is more water soluble than other corn starches, and thus, corn starch made from this waxy corn works better as an adhesive and a food thickening agent.
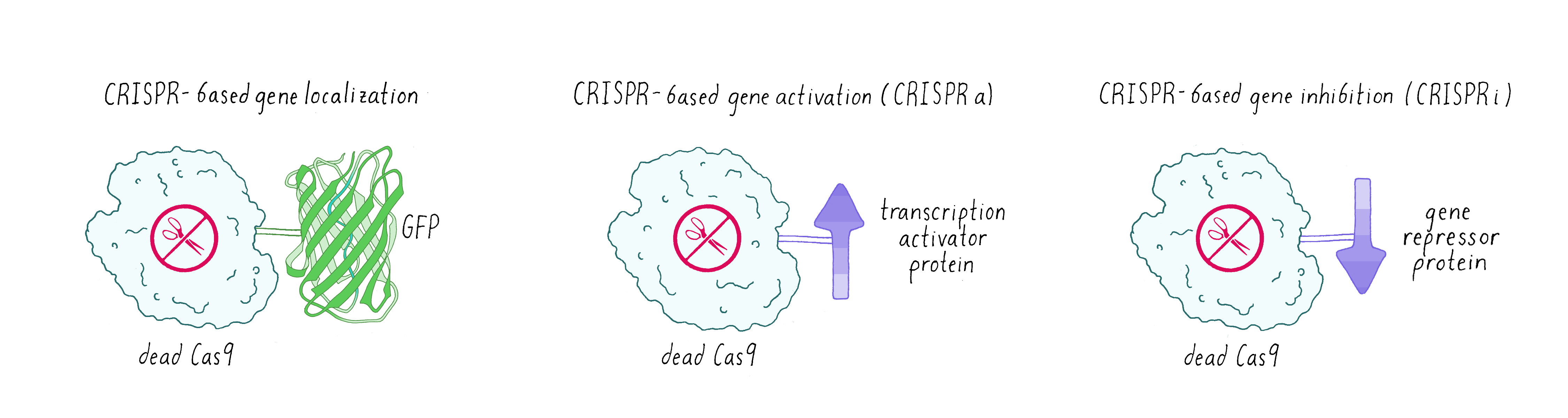
Sometimes scientists don’t want Cas9 to cut DNA. Instead, they made a mutated version of Cas9 called dead-Cas9 (dCas9), which can bind to a specific DNA sequence but cannot cut it. Scientists have tethered green fluorescent protein (GFP) to dCas9 in order to visualize where a specific gene is localized in the nucleus. Researchers have also fused dCas9 to other proteins that can either activate a gene (make more messenger RNAs and consequently the encoded protein: this is known as CRISPR-activation or CRISPRa) or turn off a gene (not make messenger RNA – CRISPR-inhibition (CRISPRi);Figure 11).
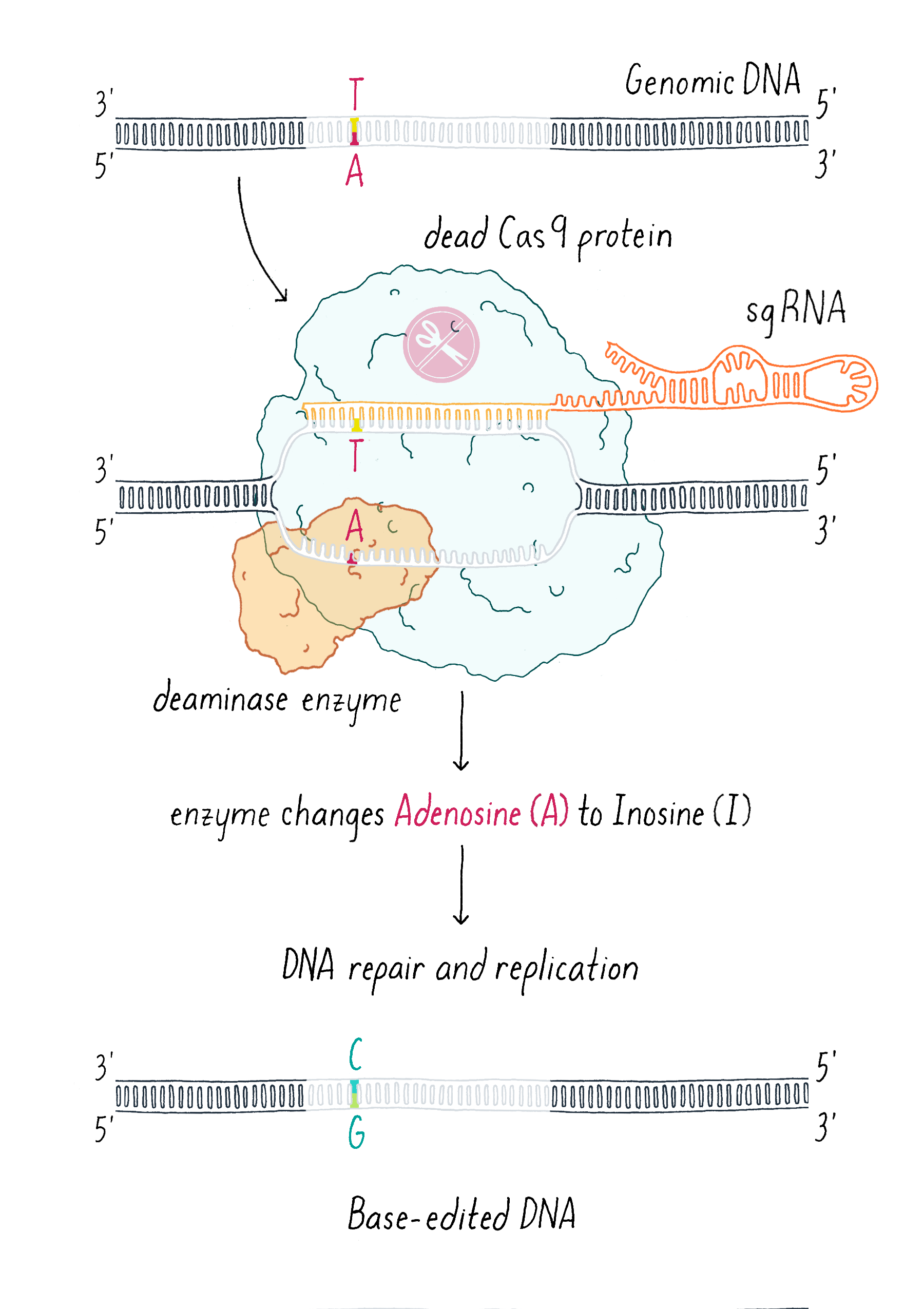
An additional clever use of dCas9 has been in DNA base editing. In this method, dCas9 is tethered to an enzyme that can chemically convert a single, specific nucleotide base into another (for example, converting an A into a G; Figure 12).
Researchers and clinicians are also actively working to use CRISPR-Cas9 to treat diseases. The goal is to program Cas9 using an sgRNA to find a specific mutation that causes a genetic disease and cut the genome at this location. The mutation can then be replaced with the DNA sequence of a healthy individual. Such a precise therapy would be ideal for curing sickle cell anemia, which is caused by one mutation in the 3 billion base pairs that comprise the human genome. Scientists and companies are actively working on trying to cure sickle cell disease with CRISPR-Cas therapy. This is now science and not science fiction.
A challenge for this type of therapy is finding a way to deliver Cas9 to the right cell or tissue. For example, if a patient has a genetic disease that affects liver cells, then Cas9 and the guide RNA should be delivered to and taken up by liver cells. Scientists are now exploring ways of delivering these molecules within the body. Another strategy for therapy is to start much earlier, either in the germline (leading to corrected egg or sperm) or in early embryos, so that the mutation is corrected in all cells in the body. While these strategies are likely to be ultimately successful, they raise ethical questions as discussed below.
Closing Thoughts
Emmanuelle and I didn’t start our collaboration with the goal of creating a new genome editing technology. We simply wanted to better understand an amazing bacterial immune system. However, in the end, our curiosity-driven research on type II CRISPR systems allowed us to engineer a simple yet powerful tool for rewriting the code of life – genomic DNA.
This experiment featured here also highlights the international and collaborative nature of science. This experiment began when I, a professor from California, met another professor, Emmanuelle, from Sweden in a café in Puerto Rico. Video conferencing technology allowed Martin and me to talk with Emmanuelle and Krzysztof at any moment to discuss our planned experiments and latest findings. This project was also driven by young scientists. In addition to Martin and Krzysztof, an undergraduate visiting UC Berkeley (Michael Hauer) played an essential role and was a co-author of this paper.
In addition to the people, the other hero of this story is Cas9, a protein that we could have never designed ourselves; instead, bacteria slowly evolved this protein over a billion years. Nature has much to teach scientists about engineering. Luck also played in that we happened to choose the Cas9 from the bacterium Streptococcus pyogenes for our experiments, which even today remains one of the most efficient Cas9 variants. Had we chosen to work with Cas9 from another species, perhaps we would have thought our experiments weren’t working, simply because they are slower and less robust Cas9 proteins.
The experiment to see if Cas9 and an sgRNA can cut a piece of DNA in a test tube has opened a Pandora’s box, the likes of which is unprecedented in the life sciences. Beyond the basic research, the use of Cas9 for genome editing has raised several environmental and ethical concerns, ones that will require conversations between scientists, ethicists, environmentalists, politicians, and general public. One major topic of debate is germline editing (edits result in a change to the sperm or egg), in which the changes made to a genome can be passed down through future generations. Should the human race tinker with its own genome and, if so, for what reasons? Most scientists and ethicists think that human genome editing could be used to correct mutations that cause horrific human diseases, such as cystic fibrosis and sickle cell anemia. But is this a slippery slope that will lead to changing other human traits and potentially alter human evolution? Another topic of active discussion is the use of genome engineering for the development of “gene drives,” which are strategies to alter the DNA of mosquitos, flies, and other disease-carrying creatures to prevent diseases. When and how should we change the genome of other species? In 2012, I never thought that I would be actively engaged in such discussions, but now I am deeply involved. Along with many other scientists and nonscientists, I feel morally obligated to help guide this new technology in responsible and useful directions.
Dig Deeper
Dig Deeper 1: The Purification of Cas9 Protein
The purification of Cas9 involved three sequential chromatographic columns. Each column focuses on a different property of the SpCas9 (affinity tag, charge, size) to isolate our protein from other unwanted macromolecules.
- The first column is called an affinity column. Cas9 is produced in E. coli with a string of six histidine amino acid residues, called a His-tag, attached to one end of Cas9. The lysate (the released content of the E. coli cells after the cells have been broken open) is mixed together with small beads that are coated with the metal nickel. Since one part of the amino acid histidine has a high affinity (binding strength) to nickel, Cas9-His-tag binds to the nickel beads. A solution with high salt is then added to the beads to wash away any macromolecules that are not bound tightly. In order to remove Cas9 from the nickel beads, a solution with the compound imidazole is washed over the beads. Imidazole binds to the nickel and displaces the histidines, so the Cas9 is detached from the beads.
- The second column is called an ion-exchange column. Since proteins are strings of amino acids and certain amino acids have a positive or negative charge at a given pH, the protein molecules themselves are often charged. The ion-exchange column uses this charged property to isolate Cas9 from other macromolecules of neutral or opposite charge. Under our conditions, Cas9 is positively charged. The beads inside the ion-exchange column are negatively charged, meaning that Cas9 will stick to the column. After we add Cas9 to the column, we run buffer over the column to remove any unwanted nucleic acid or protein. As the buffer runs over the column, we slowly raise the salt concentration in the buffer. The salt ions will then shield the charge of the column. At a certain salt concentration, Cas9 is kicked off of the negatively charged column.
- The final column is called a size-exclusion column. After running the first two columns, we are left with proteins of similar charges but different sizes. The sample is loaded onto a long column that is filled with beads containing small pores and fluid is forced through with a pump. As the proteins pass through this column, the small proteins enter inside the pores of the beads and spend time bouncing around inside of the beads as well as traveling with outside of the beads. The large proteins spend less time inside the beads or can’t fit in at all. This means that large proteins exit the column first and smaller proteins follow later. One then collects just the fractions that contain Cas9. Ideally after these three column steps, we have isolated pure Cas9 for our experiments.
References and Resources
Guided Paper
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J.A., Charpentier, E. (2012). A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337: 816–821.
This paper describes the adaptation of the bacterial immunity CRISPR-Cas system into a powerful, programmable, and easy to use tool for genome editing of any species. This research led to the Nobel prize being awarded to Doudna and Charpentier in 2020.
DownloadResources
-
CRSPR-Cas9 Video: https://www.youtube.com/watch?v=2pp17E4E-O8
A short video that gives a basic explanation of how the CRISPR-Cas9 system functions in bacteria and how scientists have adapted the system to work in other organisms.
-
Jennifer Doudna CRISPR Explanation: https://www.youtube.com/watch?v=SuAxDVBt7kQ
A video featuring Dr. Jennifer Doudna explaining how they discovered the CRISPR-Cas9 system, how it works, and the directions it is moving today.
-
Jennifer Doudna CRISPR TED Talk: https://www.youtube.com/watch?v=TdBAHexVYzc
A TED talk video that features Dr. Jennifer Doudna explaining the discovery, potential functions, and ethical issues surrounding the CRISPR-Cas9 system.
-
CRISPR FAQ: https://www.broadinstitute.org/what-broad/areas-focus/project-spotlight/questions-and-answers-about-crispr
A web page that includes a short video explaining the CRISPR-Cas9 system and has a series of frequently asked questions and their answers.
-
CRISPR-Cas9 Mechanisms and Applications: https://www.hhmi.org/biointeractive/crispr-cas-9-mechanism-applications
An interactive simulation of both how the CRISPR-Cas9 system functions and what the real-world applications are.
Activity
Learn the principles of agarose gel electrophoresis yourself and run your own simulated gels in the Activity of Gel Electrophoresis by Douglas.