Membrane Organelles
Jennifer Lippincott-Schwartz
 Jennifer Lippincott-Schwartz
Jennifer Lippincott-Schwartz
Jennifer Lippincott-Schwartz is a Senior Group Leader at the Janelia Research Campus of the Howard Hughes Medical Institute. She has pioneered the use of green fluorescent protein (GFP) technology for analyzing intracellular dynamics, including how intracellular structures form, evolve, and inter-communicate. A former President of the American Society of Cell Biology, Lippincott-Schwartz co-authored the textbook Cell Biology (with Thomas Pollard, William Earnshaw, and Graham Johnson) and is a member of the National Academy of Sciences and the National Academy of Medicine.
Summary
Eukaryotic cells house a diverse collection of internal membrane organelles such as the nucleus, ER, and Golgi, that likely enabled multicellular organisms, like us, to evolve. Organelle function is thus foundational for our current understanding of modern cell biology. So how did the field gain its present-day knowledge of organelles? The story begins with the study of secretion, the process by which proteins—such as digestive enzymes in saliva—move from the inside to the outside of a cell. Using tools of electron microscopy, genetics, and biochemistry, Nobel Prize winners George Palade, Randy Schekman, and Jim Rothman uncovered an ordered pathway by which newly synthesized proteins travel from one membrane organelle to another before exiting through the cell surface. Later, in my own laboratory and others, it was possible to directly watch this flow of material in living cells using light microscopy. Over time, a model for the molecular basis of this system was developed, which, along with other discoveries, has led to our current understanding of the nine major organelle compartments common to all eukaryotes. However, the journey is far from over. This Narrative describes not only this history but the ongoing work of engineers, computer scientists, and biologists to obtain an unprecedented high-resolution 3D view of the entire organelle system in a cell. This and other advances could offer new insights into a variety of disease states arising from disruptions of organellar function, ranging from neurodegeneration to bone disease, to viral infections.
Learning Overview
Big Concepts
Eukaryotic cells, unlike prokaryotes, have internal membrane-bounded organelles, such as the nucleus, Golgi, endoplasmic reticulum, mitochondria, as well as others. These organelles compartmentalize biochemical reactions, such as the transcription of RNA from DNA inside of the nucleus, energy production within mitochondria, or degradation within lysosomes. Small vesicles serve as carriers for transporting proteins and lipids between large membrane organelles and between these internal organelles and the plasma membrane in a process known as membrane trafficking.
Bio-Dictionary Terms Used
actin, amino acids, antibody, archaea, astrocyte, cell fractionation, chaperone, chloroplast, cholesterol, chromosome, cytoplasm, cytoskeleton, diffusion, dynein, endosymbiont, enzyme, eukaryote, exocrine cells, fluorescence microscopy, fractionation, gene, glycosylation, green fluorescent protein, GTPase, hormone, hydrophobic/hydrophilic, insulin, kinesin, lipase, lipid, microtubule, model organism, mutant, myelin, nuclease, prokaryote, protease, protein, recombinant, replica plating, ribosome, RNA, Saccharomyces cerevisiae, tissue culture, virus
Terms and Concepts Explained
autophagosome, autoradiography, coat proteins, cholesterol, electron microscopy, endoplasmic reticulum (ER), endosome, glycerophospholipid, fatty acid, focused ion beam scanning electron microscopy (FIB-SEM), Golgi apparatus, lipid droplet, lipid raft, lysosome, mitochondria, nuclear pore, nucleus, peroxisome, pulse-chase experiment, secretion, SNARE proteins, sphingolipid, sterol, temperature-sensitive mutations, tomography
Introduction
-
Eukaryotic cells are distinguished from prokaryotes by having internal membrane organelles, which provided cells with new opportunities to expand their complexity in biochemical reactions and spatial organization. Ultimately, this framework of cell organization allowed cells to organize, specialize, and work together, producing complex beings such as us.
-
Microscopy has played an important role in understanding the micrometer-sized membrane organelles inside of cells. This Narrative will discuss how technological innovations in light and electron microscopy have allowed scientists to advance our understanding of eukaryotic membrane organelles.
Part I: Journey to Discovery—How Proteins are Delivered from the Inside to the Outside of a Cell
-
Secretion is the process that produces saliva in your mouth and releases insulin from the pancreas in response to food.
-
The mechanism of how proteins were secreted from the inside of the cell to the outside was largely a mystery in 1960, largely because there were no tools to study the process.
-
Two important internal membrane structures were seen by microscopy, which provided initial clues. Camillo Golgi discovered the "Golgi apparatus" using a new staining method and light microscopy in the late 19th century. The "endoplasmic reticulum" (ER) was discovered by Keith Porter, Albert Claude, and colleagues using electron microscopy in the 1960s.
-
An important breakthrough experiment was performed in the 1960s by Palade and Jamieson. They performed a "pulse chase experiment" in which a radioactive amino acid was given to cells for a short period of time and then removed. Newly synthesized proteins incorporated the radioactivity and the location of the radioactively labeled proteins was determined by autoradiography and electron microscopy. They also performed cell fractionation studies to confirm their findings.
-
The Jamieson-Palade experiment revealed a pathway for secretion. Proteins appear first in the ER, then in the Golgi, and then in secretory vesicles in the cytoplasm.
-
The experiments of Palade and Jamieson provided a pathway for secretion through successive membrane compartments. But the molecules involved were unknown. Work by Randy Schekman and James Rothman took the problem of protein secretion to a molecular level.
-
Schekman and his colleagues used a genetic approach in the baker's yeast Saccharomyces cerevisiae to find genes involved in secretion. As genes involved in secretion are likely essential for life, he employed a specific screen to look for "temperature-sensitive" mutations.
-
Schekman found a mutation in a gene, called sec-1, that blocked the secretion of an enzyme when the yeast was grown at a higher temperature but not at a lower temperature. Remarkably, when examined by EM at higher temperature, the yeast cells were filled with small vesicles, which were normally transient intermediates in the secretion pathway but accumulated at high numbers due to the mutation.
-
James Rothman and colleagues used a biochemical approach in mammalian cells to understand how secretory proteins pass through the Golgi apparatus. They developed a reconstitution assay in which they could detect if a protein (VSV G protein from a virus) passed from one Golgi compartment (cis-Golgi) to another (medial-Golgi).
-
The assay made use of extracts from two types of cells. One cell type was infected with a virus that made the VSV G protein but lacked an enzyme that transferred a sugar to the G protein. The second cell type had the normal sugar transferase enzyme but lacked virus and VSV G protein. When extracts from the two cell types were mixed, the sugar was transferred onto the G protein, because the VSV G protein in the cis-Golgi from cell type 1 was transferred to the medial-Golgi from cell type 2.
-
The genetic screens by Schekman and the biochemical screens by Rothman resulted in the discovery of many proteins involved in secretion. Many of these proteins are conserved from yeast to mammals.
-
The author (Jennifer Lippincott-Schwartz) attached a green fluorescent protein (GFP) to the VSV G protein and, using live cell microscopy, was able to make movies of the entire secretory process from the ER to plasma membrane .
-
Lippincott-Schwartz also found that a drug called brefeldin A made the Golgi apparatus rapidly disappear and assimilate into the ER. This indicated that the Golgi is not a "permanent" structure, as was previously thought. It also indicated that the secretion is not just a one-way pathway and rather involves bi-directional exchange between membrane compartments (e.g., bidirectional trafficking between the ER and the Golgi).
Part II: Knowledge Overview—The Internal Membrane Systems of Eukaryotic Cells
-
Membrane organelles play roles in 1) uptake of molecules from outside the cell, 2) synthesis, processing and transport of proteins, 3) energy production, and 4) housing the genetic material.
-
Eukaryotic cell membranes are composed of fluid lipid bilayers with embedded and peripherally associated proteins that are highly dynamic.
-
There are three major classes of lipids in eukaryotic membranes: 1) glycerophospholipid, 2) sphingolipids, and 3) sterols.
-
Sphingolipids, cholesterol, and certain protein can be organized into small, separate domains in the membrane called "lipid rafts."
-
There are nine membrane-bound organelles that are found throughout eukaryotes: 1) nucleus, 2) endoplasmic reticulum, 3) Golgi apparatus, 4) endosome, 5) autophagosome, 6) lysosome, 7) mitochondria, 8) lipid droplet, and 9) peroxisome. Plants also have chloroplasts.
-
The nucleus contains the genetic material (DNA) and has nuclear pores that allow proteins and RNA to pass through.
-
The endoplasmic reticulum is where ribosomes dock and insert proteins into the membrane. It is also an important site for lipid processing and calcium storage.
-
The Golgi apparatus, a series of flattened cisternae, modifies proteins and lipids, and sorts them for further transport to other membrane compartments in the cell.
-
Endosomes are internal membranes that have budded off from the plasma membrane and can contain materials derived from the outside of the cell (e.g., hormones, nutrients).
-
Lysosomes are the major digestive organelle of the cell.
-
Autophagosomes are double-membraned structures that engulf portions of the cytoplasm or organelles and deliver them to the lysosome for degradation.
-
Peroxisomes house enzymes that convert long chain fatty acids and branched fatty acids into simpler substrates for use by mitochondria in energy production.
-
Lipid droplets, which are surrounded by a lipid monolayer rather than bilayer, store lipids and cholesterol and serve as a reservoir for making membranes or energy production.
-
Mitochondria are the main energy production organelles of the cell. Mitochondria have a double membrane (inner and outer membranes) and possess a small DNA genome.
-
Different organelles have distinct lipid compositions that help to define their identity and function. In the secretory pathway, the percentage of sterol/sphingolipid is low in the ER and high in the plasma membrane.
-
A variety of proteins aid organelle function and activities, including 1) defining the shape of an organelle, 2) transporting membranes along cytoskeletal elements, 3) tethering different organelles to each other, and 4) directing the budding and fusion of membrane vesicles.
-
Membrane vesicles act as carriers of proteins and lipids between large membrane compartments. Coat proteins promote vesicle budding from one membrane compartment; the coat is removed after the vesicle is formed, and then the vesicle fuses with another membrane compartment through the actions of tethering and SNARE proteins.
-
Tethering proteins can bring different large membrane organelles very close to allow exchange of calcium and lipids.
-
The initial internal membrane compartment of eukaryotic cells is thought to have arisen from a fusion between an archaea host cell and a bacterium, with the bacteria endosymbiont evolving into mitochondrion. A later fusion event with a cyanobacterium may have given rise to chloroplasts.
Part III: Frontiers—A Return to Electron Microscopy for Visualizing Membrane Organelles in 3D
-
Exciting new EM technology is emerging to visualize all internal membrane organelles in a 3D view at high resolution.
-
A single EM image provides a 2D image, effectively a slice of a 3D cell.
-
Many sequential 2D images through a cell can be assembled computationally into a 3D rendering, a process known as tomography.
-
One method for achieving a series of sequential 2D image slices through a 3D sample is called focused ion bean scanning electron microscopy (FIB-SEM).
-
To prepare the biological sample, the material is "fixed," dehydrated (water removed), stained with osmium to highlight biological structures, and embedded in a plastic resin.
-
The plastic block with the cell inside is then put in the FIB-SEM machine. The machine alternates between milling the sample (removing 2–8 nanometers from the plastic block) and then imaging the surface of the block.
-
To create a 3D image, the FIB-SEM microscope operates through alternating imaging and milling cycles.
-
Finally, all of the slices are assembled into a 3D image. The image then has to be interpreted by computers to identify outlines of membrane organelles and other structures. This challenging process requires the skills of computer scientists who write algorithms that allow computers to recognize features in images.
-
In addition to understanding the organization of membrane organelles in cells, FIB-SEM is being applied to larger structures, for example, determining all of the connections between neurons in a brain.
Closing Thoughts
-
The dysfunction of membrane organelles is associated with many human diseases.
-
New technologies for measuring the activities of membrane organelles in tissues might help to develop new therapeutic strategies for treating disease.
Guided Paper
Novick, P. and Schekman, R. Secretion and cell-surface growth are blocked in a temperature-sensitive mutant of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1979;76:1858–1862.
A classic paper on yeast genetics describes finding genes involved in protein secretion for the first time. This research led to Schekman receiving his Nobel prize in 2013.
DownloadIntroduction
What allowed human beings to become more complex than bacteria? Let us look back about two billion years ago for a possible explanation.
For the first 1.5 billion years of life on earth (3.5 to 2 billion years ago), prokaryotes ruled the earth. Prokaryotes were (and are) small and relatively simple in their structure, with just a boundary membrane. There were two types of prokaryotes both then and now—archaea and bacteria. Then, around two billion years ago, an evolutionary event may have occurred that changed the game of life on the planet. We do not know the full details, but it is speculated that an archaea swallowed a bacterium. Like the story of Jonah and the whale, the swallowed bacteria survived and adapted to its new role inside the archaea. The relationship between the host and its internal endosymbiont evolved over time, ultimately resulting in a new type of cell with an elaborate internal membrane system. That cell type, to which our own cells belong, is the eukaryote.
There are now many different eukaryote cell types spanning from yeast to human, yet they all share similar internal architectures. Eukaryotic cells—making up all plants and animals on earth—contain nine different membrane organelles. These are the nucleus, the endoplasmic reticulum (ER), Golgi apparatus, endosome, lysosome, autophagosome, lipid droplet, peroxisome, and mitochondria (and, in addition, chloroplasts in plants). In this Narrative, we explore what these organelles do and why they are necessary for eukaryotic life on earth.
One way to think of a cell is as a chemical factory, with reaction vessels and pipelines designed to optimize the synthesis of specific chemical compounds or processes. The membrane organelles are the reaction vessels and their molecular communication networks are the pipelines. This interlinked system is optimized to allow cells to organize, specialize, and work together, building the larger systems of the body such as tissues, organs, and the circulatory and nervous systems necessary for complex life.
How we have come to understand the workings of this chemical factory is an amazing story. It started with biologists realizing that cells have to communicate with the outside world to survive. Not only do cells need to take up materials to grow and divide but also they need to release proteins through their plasma membrane to make their environment habitable and compatible with the cell's growth and activities. Examples include the release of insulin from the pancreas or the secretion of digestive enzymes into saliva. Researchers speculated that there were internal cellular compartments (i.e., organelles) specialized for this activity. But demonstrating the existence of such organelles was difficult until technologies, such as electron microscopy, allowed researchers to visualize and study these membrane organelles.
As we will learn, the discovery of eukaryotic organelles and their membrane trafficking pathways was driven by creative scientists who employed new approaches and technologies. What was revealed through their collaborative efforts has profoundly affected our understanding of how organelles operate as functional chemical units inside cells. These organelles also play important roles in disease. As just one example, pandemic-causing viruses, like coronavirus Covid-19, hijack the same membrane organelle pathways specialized for digestion and secretion. The Covid-19 virus gets inside the cell through a process called endocytosis in which it is wrapped in cellular membranes. After making more copies of itself, the viral genome enwraps itself in ER/Golgi membranes to reform a viral particle. These particles then follow the secretory pathway (similar to digestive enzymes) for their release outside the cell, where they find other cells to infect. Understanding how Covid-19 hijacks membrane organelles could help to devise strategies to prevent the propagation of this and many other viruses.
In the Journey to Discovery, we will explore the experiments that led to our modern understanding of the secretory pathway and its constituent organelles. This Nobel prize-filled journey begins with George Palade, often referred to as the "father of modern cell biology," who used electron microscopy (EM) as a key tool to understand membrane organelles and how proteins flow through them. What Palade found was that secretory products pass through a series of membrane compartments (the ER and the Golgi) en route to the plasma membrane. These results, acquired in the 1960s and 1970s, set the agenda for work in the field for the coming decades. Palade's work stimulated Randy Schekman and Jim Rothman to devise genetic and biochemical methods to identify the molecules that enable the trafficking of proteins through organelles in the secretory pathway. In the Knowledge Overview, we discuss some of these molecules as well as survey the identities and functions of the nine internal membrane compartments of the cell and how they communicate with each other. In the Frontiers section, I will take you to the leading edge of my own work at the Janelia Research Campus. We are using a new type of EM (with the long name of "focused ion beam scanning electron microscopy") to produce very high resolution 3D views of all membranes in a cell. In the Closing Thoughts, I will share some thoughts on how knowing more about membrane organelles might allow us to better treat or prevent disease.
Beyond learning more about membrane organelles, I also hope to convey how technology and assay development drives progress in science. The Nobel Prize work of George Palade was made possible by the development of the electron microscope (EM) and its application to the study of cells. The high-resolution images of EM allowed scientists to see new things inside of cells, which were not visible by light microscopy. Now, EM is again having an exciting new renaissance. Many advances are being made by biologists, physicists, engineers, chemists, and computer scientists through the use of enhanced EM techniques to see more details of intracellular processes. The progress being made in understanding membrane organelles is staggering. I hope to share some of this excitement with you as well.
Part I: Journey to Discovery —
How Proteins Are Delivered From the Inside to the Outside of A Cell
The Problem
Have you ever wondered how your mouth (or your dog's mouth; Figure 1) waters at the sight of food? The answer to this question involves a fundamental process inside cells called secretion. Secretion is what produces saliva and allows the pancreas to release insulin in response to food. During secretion, proteins are delivered from the inside of the cell to the outside. For dogs, secretion of saliva, which is loaded with digestive enzymes, allows food to begin being broken down in the dog's mouth before reaching the stomach.
Ivan Pavlov, a scientist from Russia, was one of the first researchers to describe secretion, winning the Nobel Prize in Physiology or Medicine in 1904 for his work on the physiology of digestion. By isolating a salivary gland from a dog, he observed that the amount of saliva increased and changed its composition in response to different foods. He even noticed that dogs could be trained to salivate before food was presented.
Where does extra saliva come from when food is sensed or tasted? Pavlov's work demonstrated that it derives from the ability of cells in the salivary gland to produce secretory products, in this case food-digesting enzymes. But this process is not unique to the salivary gland. Secretion occurs in all cells and is critical for many aspects of cell function. In addition to allowing cells to release a myriad of newly synthesized molecules, the secretory process enables the cell surface to be decorated with proteins involved in cel-to-cell communication and the uptake of extracellular components.
How a eukaryotic cell can secrete proteins was largely a mystery until the mid-1960s. Three barriers prevented scientists from understanding the secretory pathway of cells:
1. A lack of tools to visualize cells and their internal compartments.
2. Methods to tag and follow how proteins move from one location to another inside of cells.
3. Strategies to find the genes involved in secretion and characterize their encoded proteins.
As this Journey to Discovery unfolds, we will see the importance of developing new techniques for answering questions in biology. New tools allow scientists to see new things and do experiments that previously were not possible.
Two key players will be introduced in the first clues: the endoplasmic reticulum (ER) and the Golgi apparatus.
Clues
Clue 1: The initial observations of the Golgi apparatus and endoplasmic reticulum
A vital tool that provided an impetus for studying the secretory pathway came from the work of Camillo Golgi, an Italian biologist interested in the nervous system. Brain organization at the cellular level was difficult to study during Golgi's time, because cells were hard to identify. Microscopes back then were not very powerful, and the contents of cells were largely invisible. To overcome this problem, Golgi developed a staining technique in the late 19th century that produced a black reaction product that labeled the cell's outer membrane and interior compartments. Golgi used this technique to identify structures of the nervous system, and he and the Spanish scientist Ramon y Cajal were awarded the Nobel Prize in 1906 for their independent work.
Golgi also noticed thread-like networks inside of cells of unknown function (Figure 2). The term "Golgi apparatus" was used in 1910 to describe this structure and first appeared in the scientific literature in 1913. However, without an obvious cellular role, some scientists argued these networks were staining artifacts, while others ignored them altogether. This view persisted until the early 1960s.
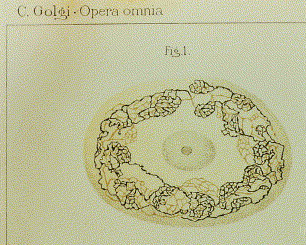
Much later (in the late 1960s), using electron microscopy, French and Canadian scientists Alain Rambourg and C. Leblond saw internal network structures similar to those previously reported by Golgi. Seeing the same structure using different methods convinced many scientists that the Golgi apparatus was real. What was the function of the Golgi apparatus? The answer was not obvious from the images.
The endoplasmic reticulum (or ER) was first observed with light microscope over a century ago but was more definitely characterized by electron microscopy by Keith Porter, Albert Claude, and colleagues (Figure 3). Later, the word "reticulum," which means "network," was coined to describe this lattice of membranes. However, the function of the endoplasmic reticulum again was not obvious from the images alone.
Clue 2: George Palade finds a secretory pathway that moves proteins between compartments
It was not until the 1960s with the work of George Palade and colleagues that the roles of the ER and Golgi in secretion became clear.
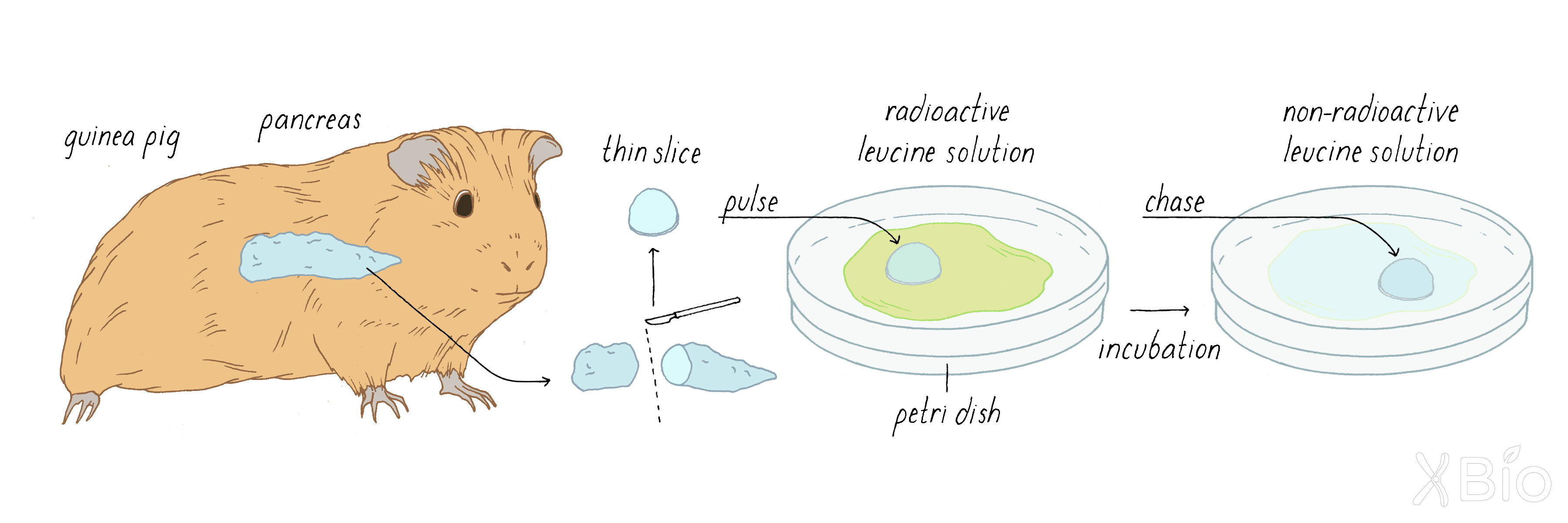
Palade decided to study the pancreas of a guinea pig as a model system, because this organ is highly specialized for secretion. Exocrine cells of the pancreas secrete digestive enzymes that help to breakdown food. Because of this highly specialized organ function, the majority of proteins being produced by pancreatic exocrine cells are secretory enzymes.
Explorer's Question: What other cell type might be particularly good for studying secretion?
A.Epidermis skin cell
B.Heart muscle cell
C.Kidney cell
D.Lung cells
E.Neuron in the brain
Answer: D. All eukaryotic cells secrete some amount of protein, so it could be studied in any cell. However, Palade choose pancreatic cells, because these cells are highly specialized for secretion, which made it easier for him to study this process. Cells in the lung also secrete a substance called mucus, which is particularly prominent when you have a cold.
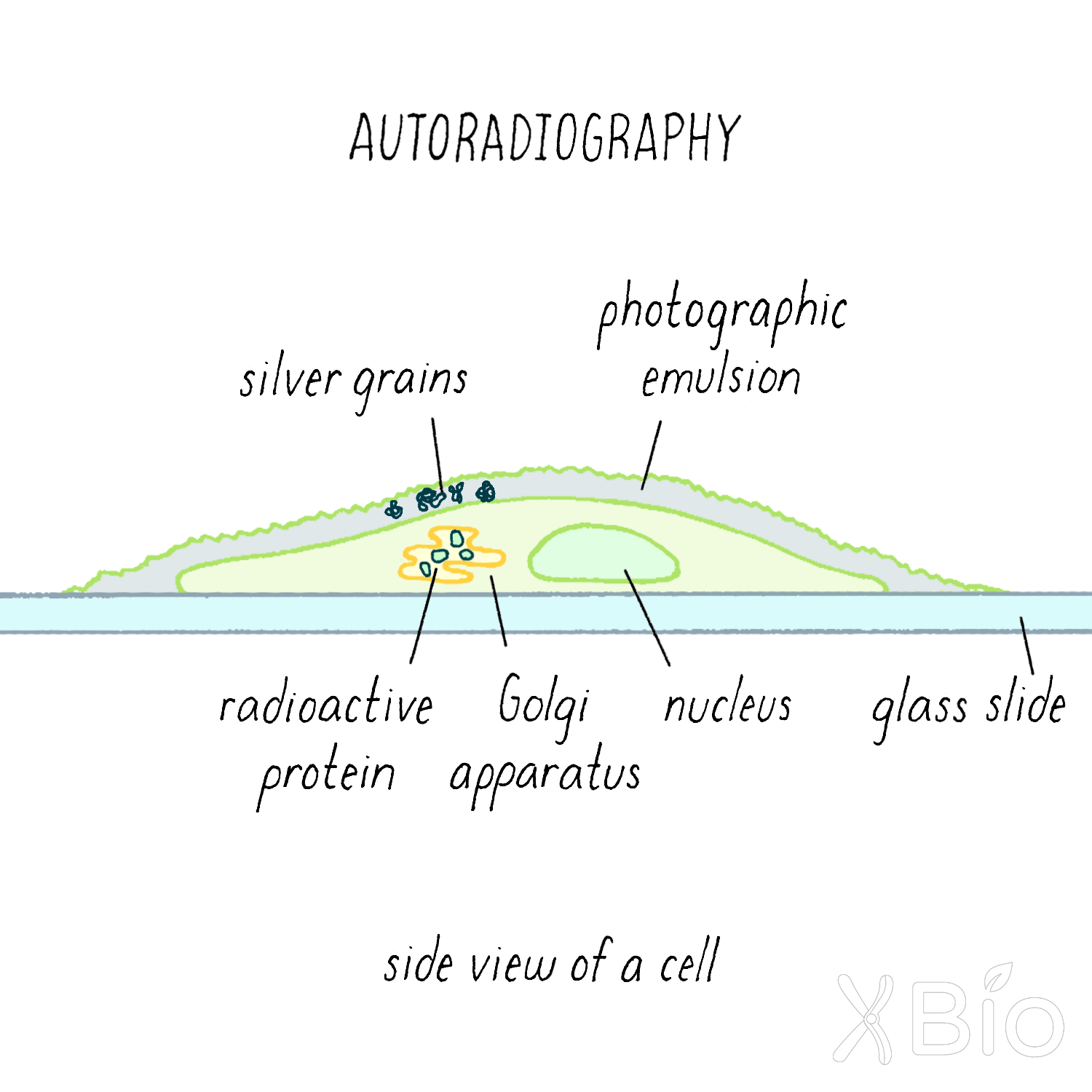
Palade helped to develop two new techniques that were crucial for making progress on understanding secretion: 1) electron microscopy (EM) autoradiography and 2) cell fractionation.
EM autoradiography involves exposing an X-ray film to the decay emissions of a radioactive substance (Figure 4). The film is placed directly next to the radioactively labeled tissue section, which is covered with a thin layer of emulsion to trap the radiographic signal. In the X-ray image, the sites of the labeled substance appear as black, autoradiographic grains.
To study the secretory pathway using autoradiography, Palade injected the radioactive amino acid leucine into a guinea pig. The radioactive leucine was quickly taken up by cells in the guinea pig, including the exocrine cells of the pancreas. Amino acids are building blocks of proteins and the introduced radioactive leucine became incorporated into newly synthesized proteins for a short while, until the bolus of injected radioactive leucine disappeared by excretion. If Palade sacrificed the guinea pigs shortly after injecting radioactive leucine and used autoradiography to visualize proteins with radioactive leucine, he could determine where newly synthesized radioactive proteins are first made in the cell. If he waited longer after injection, then he could determine how newly synthesized proteins travel from the inside to the outside of the cell over time. This type of experiment is called a "pulse-chase experiment."
Jamieson and Palade later found that they could deliver a more precisely timed pulse-chase by taking the pancreas out of the guinea pig and slicing it and then applying radioactive leucine for a short time (the slices still continued to make and secrete proteins). By using this approach (Figure 5), Jamieson and Palade could trace the intracellular pathway of these proteins.
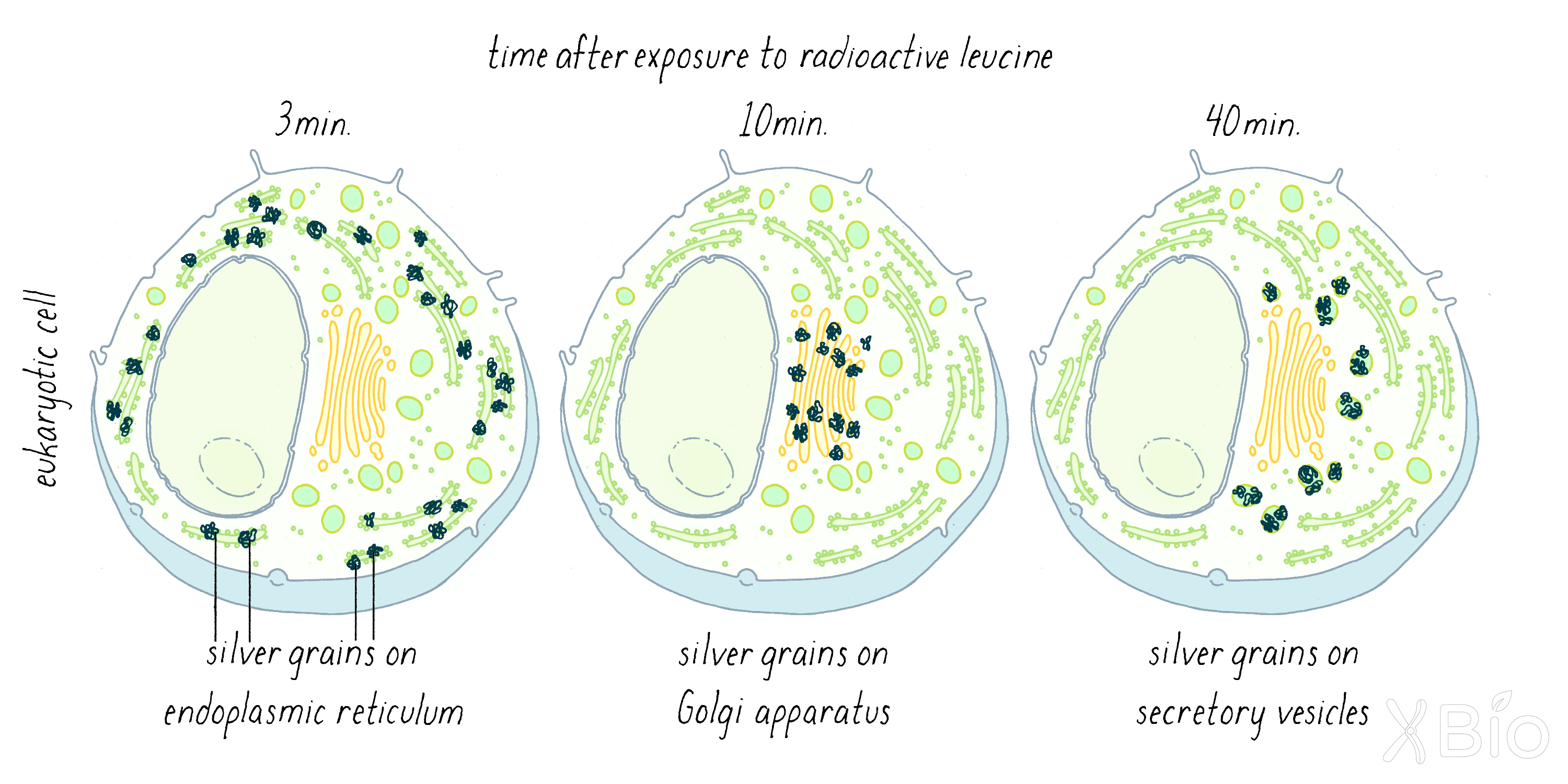
Palade's autoradiographic observations showed that at ˜3 minutes after injection, the labeled proteins were localized mostly at "rough surfaced elements" of the ER (Figure 6; left). The rough features are ribosomes, which synthesize proteins and insert them into the interior of the ER. At ˜10 min, the grains appeared in the Golgi apparatus (Figure 6; middle). After 40 minutes, the grains were seen in membrane vesicles called secretory vesicles (Figure 6; right). This work showed convincingly that secretory proteins appeared first in the ER, then moved to the Golgi apparatus, and then exited the Golgi in secretory vesicles that ultimately are delivered to the plasma membrane.
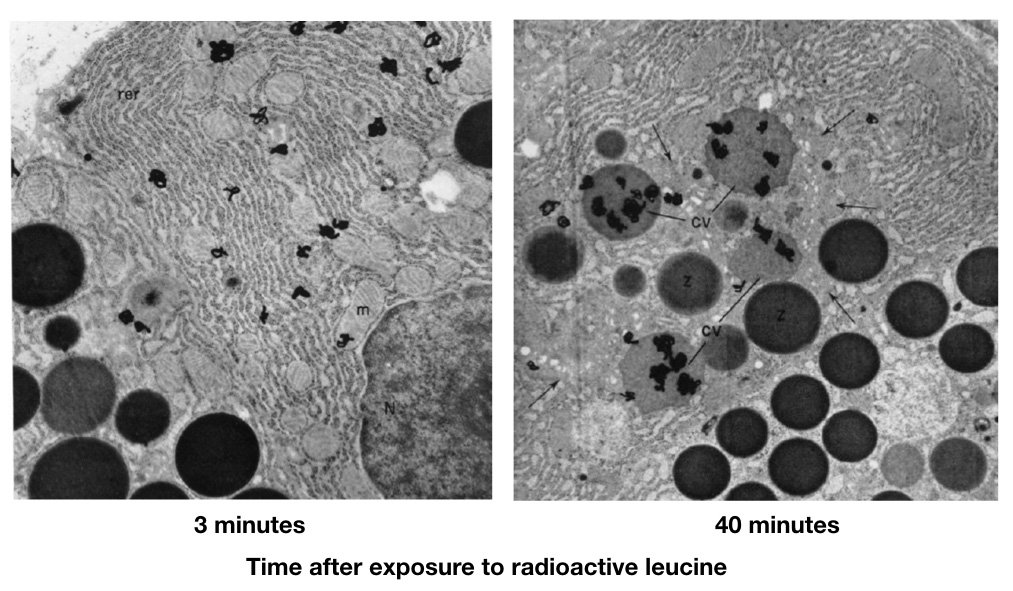
A subset of the real data from the Jamieson and Palade experiment is shown below (the 3-min and 40-min time points; Figure 7).
Jamieson and Palade also used a biochemical approach called "cell fractionation" to confirm their results and conclusion by another method. Cell fractionation involves gently breaking open cells in buffered solution so that the cell's various organelles remain intact. The extract is then centrifuged through a solution gradient of increasing sucrose concentration. As different organelles have varying sizes and densities (i.e.,isolated ER membranes are heavier than isolated Golgi or plasma membrane membranes), they segregate from each other or "fractionate" during centrifugation.
To apply cell fractionation to his studies of the secretory pathway, Jamieson and Palade added radioactive leucine to the slice of pancreas, chased with a solution without the radioactive leucine for different times, and then performed cell fractionation to see where the radiolabeled proteins were found. At early times, the radioactivity was found mostly in the fraction enriched for ER, but at later times, radioactivity was found in the fractions containing the Golgi and at still later times in fractions containing mature secretory granules.
Palade's complementary morphological and biochemical data provided powerful support for a model of vectorial transport of secretory proteins through cells, with transport through the Golgi apparatus being a key feature (Figure 8). The work suggested that traffic to the final plasma membrane destination was mediated by smaller membrane vesicles that carry proteins between the larger membrane compartments.
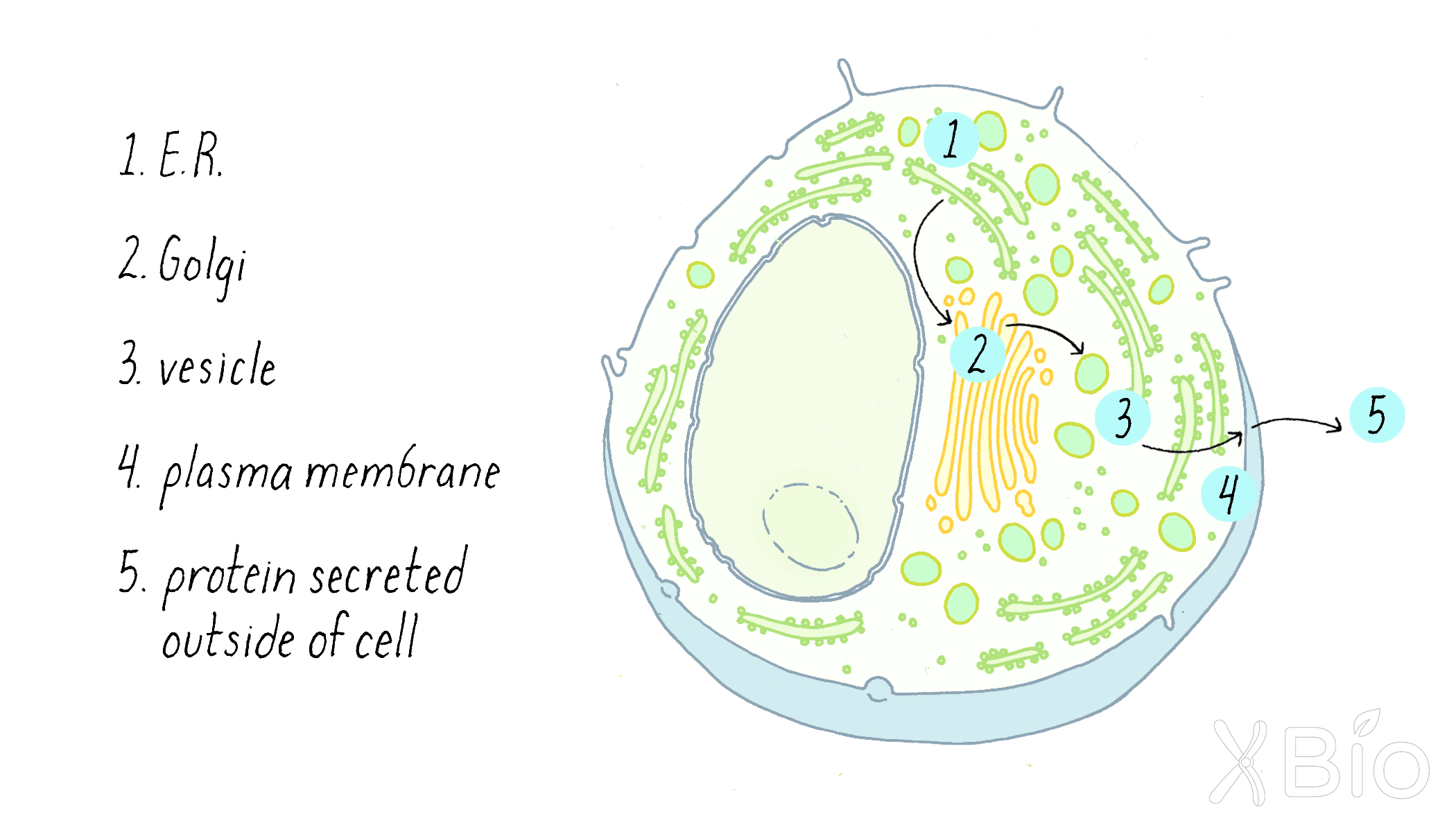
The Palade model for membrane secretion set the agenda for work in the field for the coming decades. The key next questions revolved around identifying the molecular machinery that moved proteins from one membrane compartment to the next in the secretory pathway, as will be described next.
The Discovery: Molecules involved in protein secretion
Finding the machinery involved in the secretory pathway was made possible by genetic studies in the laboratories of Randy Schekman and biochemical studies in the laboratory of Jim Rothman. Their story reveals how these complementary approaches can be used to identify genes and their encoded proteins that are involved in biological processes. Rothman and Schekman were awarded the Nobel Prize in Physiology or Medicine in 2013 for their discoveries.
Discovery Part A: Genetics uncovers proteins involved in membrane trafficking
Simple yeast cells (the type used for baking bread; also called Saccharomyces cerevisiae) secrete certain proteins. Randy Schekman (Video 1) decided to study yeast secretion with the belief that the secretion process was basic and universal and that whatever he learned in yeast would be applicable to humans (and he was correct). The reason for choosing yeast is because it is a powerful model organism for genetic studies. Schekman hoped to use genetic approaches to identify genes involved in secretion.
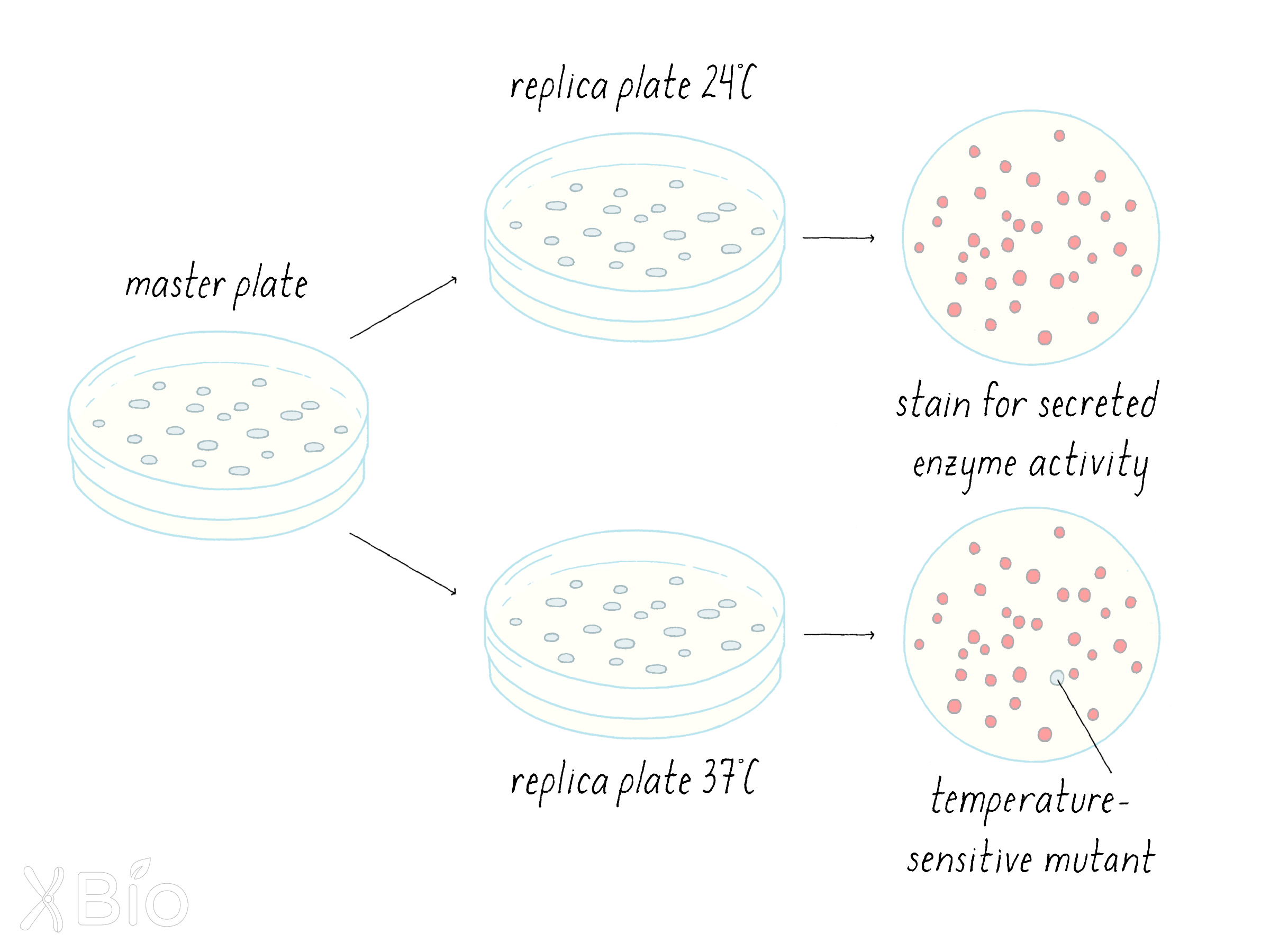
Schekman and his graduate student Peter Novick decided to look for mutants in yeast that interfered with protein secretion (see the Guided Paper). A mutation is a change to the DNA sequence of a gene, which can result in the production of a defective protein. The one problem with having a serious mutation in secretion is that it would likely kill the yeast cells, or at minimum prevent them from growing. If a mutant yeast cannot grow and divide, it also cannot be easily studied. To get around this problem, Schekman looked for a special kind of mutation—a temperature-sensitive (ts) mutation (Dig Deeper 1 to learn more about temperature-sensitive mutations). At one temperature (the "permissive temperature"; 24°C), the effect of the mutation is mild; secretion occurs, and the yeast can live and propagate. However, at a higher temperature (the "restrictive temperature"; 37°C), the effect of the mutation is severe; secretion is blocked; and the yeast will not grow.
Schekman first looked for temperature-sensitive lethal mutations. On a petri plate with growth agar, he grew up many colonies of yeast, each derived from a single cell; thus all cells in a colony are clones of one another. The colonies were transferred from the original plate onto replicate plates, so that the colonies would be positioned in the same relative geographic positions. One plate was grown at 24°C and its replicate partner plate was grown at 37°C. Schekman looked for colonies that grew at 24°C but were lethal at 37°C. He found 87 such temperature-sensitive yeast colonies out of a total of 1600.
Explorer's Question: Do you think that all 87 mutants were interesting to study as a way of understanding the mechanism of secretion?
Answer: No. The temperature-sensitive clones might die at 37°C for many reasons, not necessarily a defect in secretion.
Next, the 87 clonal yeast were examined for ones that might have a secretion defect. Again, he made replicate plates and now tested the 87 colonies for a secreted enzyme (a phosphatase) whose activity could be detected with a staining procedure (Figure 9). Schekman was looking for a rare mutant cell that would secrete enzymes at 24°C but not 37°C. He found a single one out of the 87 clones and named it sec1.
Next, Schekman measured secretion over time in a more quantitative manner (Figure 10).
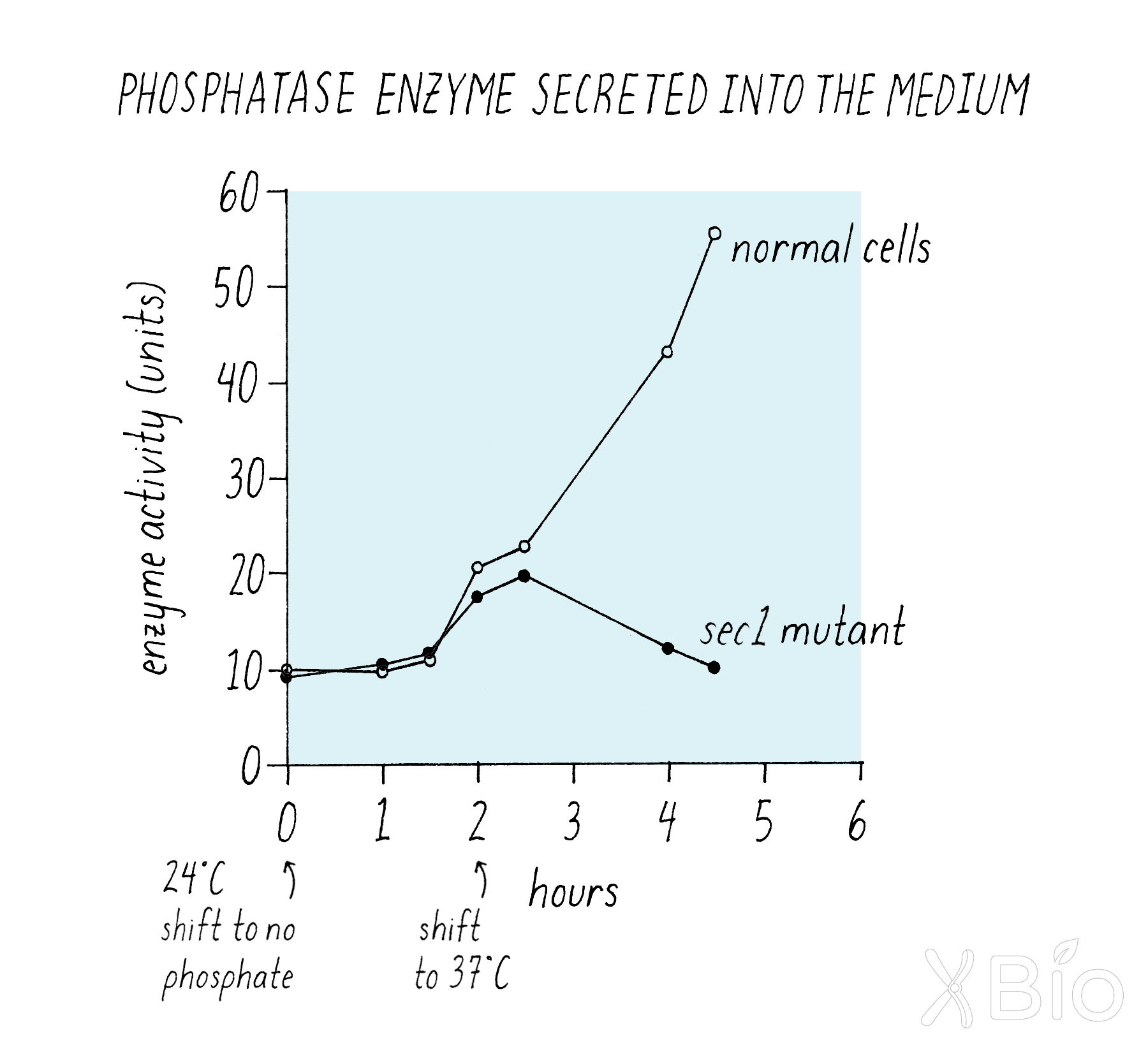
Explorer's Question: What does this experiment tell you about protein secretion by the sec1 mutant at 24°C and 37°C?
Answer: The wild type and mutant yeast both secreted the enzyme at equal rates at 24°C, but when the temperature was shifted to 37°C, the secretion into the medium halted for the sec1 mutant cells, but not for the wild type yeast (Figure 10).
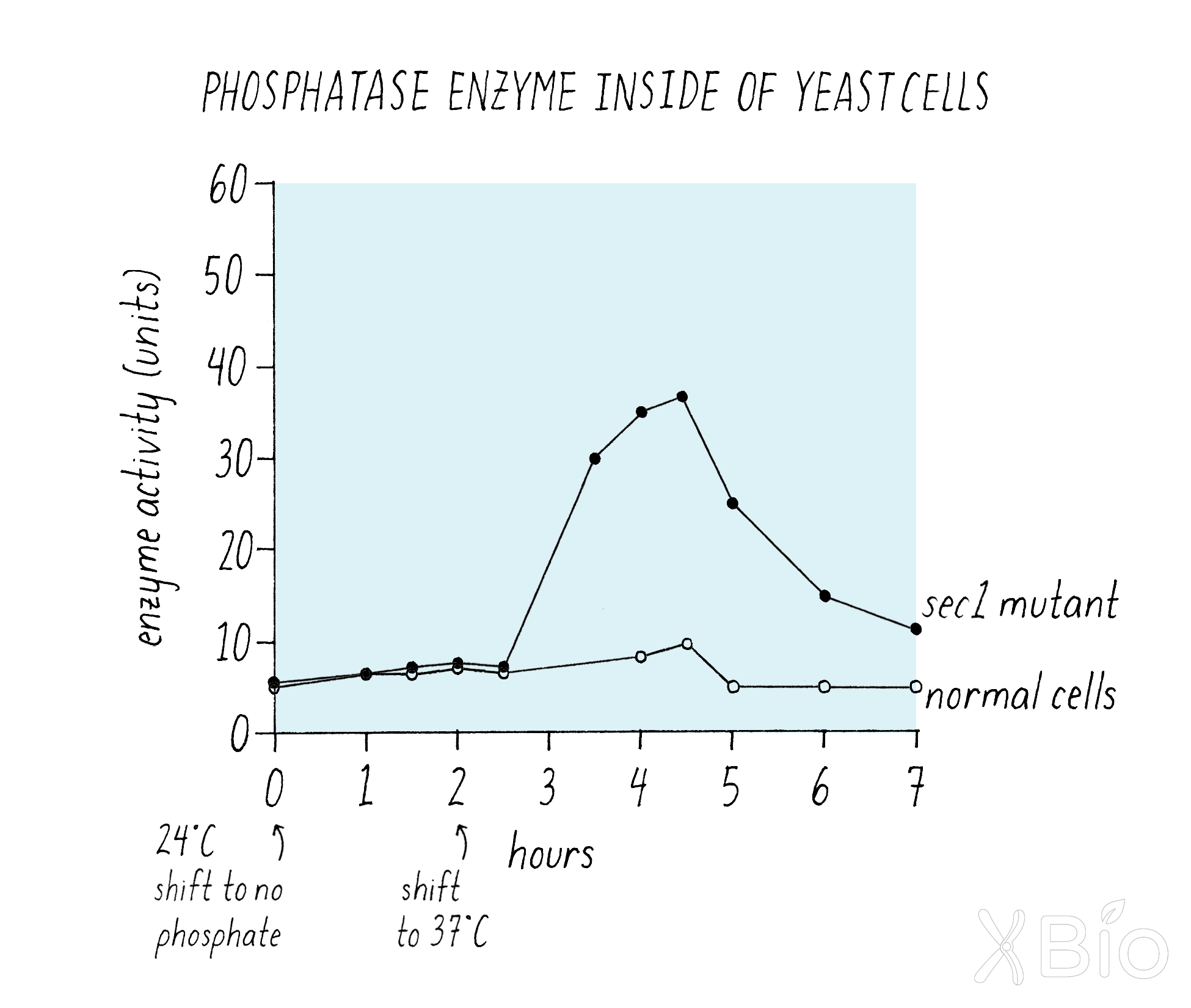
If the enzyme was not getting into the medium, one might expect that it is being trapped inside of the cell. Indeed, Schekman showed that this was the case (Figure 11). Wild type yeast cells retain very little phosphatase enzyme both at 24°C and 37°C, because the enzyme moves efficiently from its synthesis through the secretion pathway to the medium. However, the mutant cells suddenly started accumulating enzyme inside once the temperature was shifted to 37°C (Figure 11).
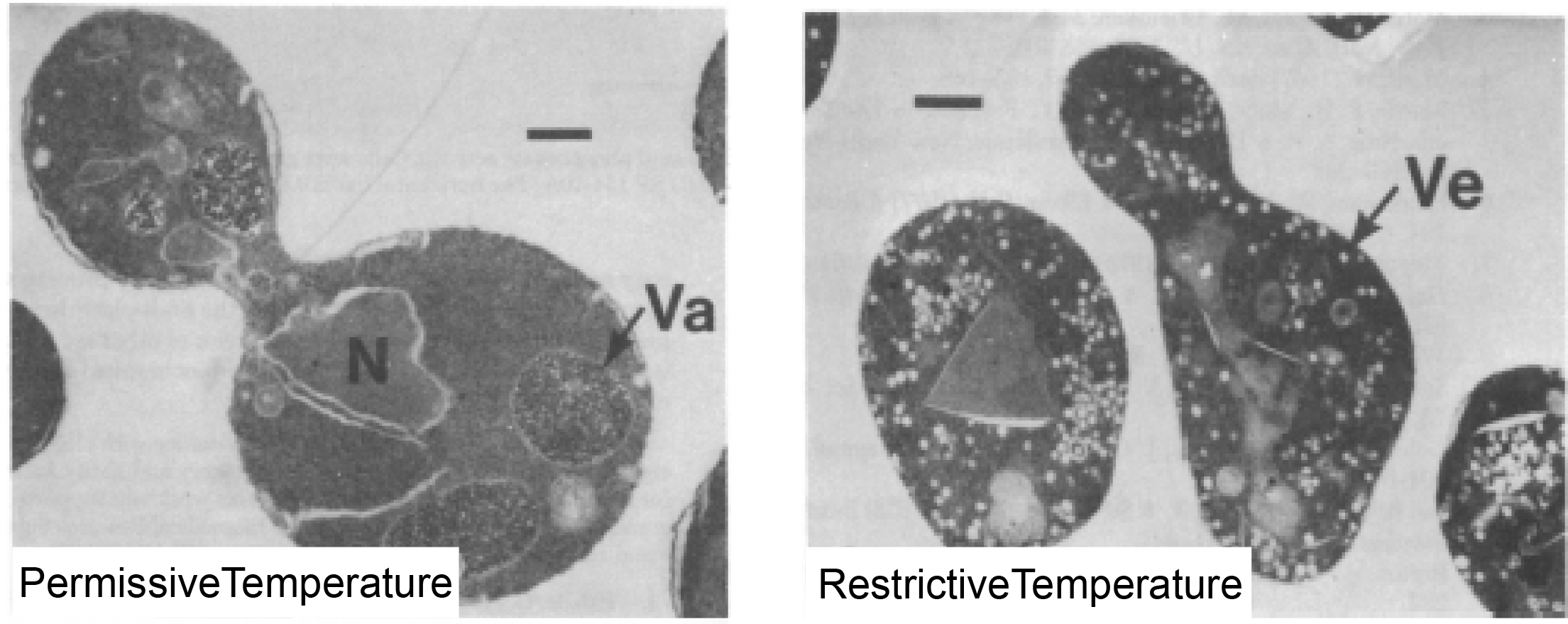
Next, following the footsteps of Palade, Schekman looked at the sec1 mutant yeast by electron microscopy to see if it was possible to spot a problem visually. What he saw was remarkable (Figure 12). At the permissive temperature, the sec1 cells looked fairly normal. But at the restrictive temperature, the cells were packed full of small vesicles.
Explorer's Question:Why do you think that the sec1 cell is packed with small vesicles?
Answer: Presumably, these vesicles were on their way from one destination to another in the secretion pathway but were stopped short of reaching their final destination as a consequence of the sec1 gene mutation. As a result, these vesicles, which are normally transitory, began to accumulate.
Schekman went on to identify numerous other "sec" mutants. All told, Schekman's approach identified more than 50 genes involved in the secretory pathway. The different genes were involved in many different steps in secretion, essentially creating a molecular map from start (ER) to finish (fusing with the plasma membrane). This map proved similar to that charted by Palade for the secretion of proteins in pancreatic cells. Schekman's work mechanistically confirmed and extended Palade's view that small vesicles derived from a preexisting organelle move proteins from one organelle to another and then to the plasma membrane in the secretory pathway. We will describe the functions of some of these proteins in the Knowledge Overview.
Discovery Part B: Biochemistry reveals molecules involved in vesicular trafficking in the Golgi
James Rothman (Video 2) decided to use a biochemical approach to understand how secretory proteins pass through the Golgi apparatus. The Golgi apparatus itself is composed of three sub-compartments (cis, medial, and trans Golgi) that proteins pass through on their way to secretion. The cis Golgi receives proteins from the ER via small vesicles, then passes those proteins sequentially through the medial and trans Golgi, and finally the trans Golgi releases the processed proteins in vesicles for delivery to the cell surface. The Rothman developed a "test tube" (in vitro) reconstitution assay that measured how proteins pass from the "cis" Golgi compartment to the "medial" Golgi compartment in mammalian cells.
How can one measure biochemically whether a protein has passed from one Golgi compartment to another? Rothman knew that sugars are added to proteins as they traverse through the Golgi. The addition of a sugar group to a protein is called "glycosylation." A specific type of sugar (N-acetyl-glucosamine) is added only in medial-Golgi (Figure 13, left). By putting a radioactive label on this sugar, they could detect if it got added to a protein as it passes through the Golgi. Rothman knew that when some viruses infect an animal cell, they take over the cell's protein-making machinery, forcing the cell to make large amounts of viral protein. Taking advantage of this trick, Rothman infected mammalian cells with vesicular stomatitis virus and followed the addition of a sugar to one viral protein—the VSV G protein.
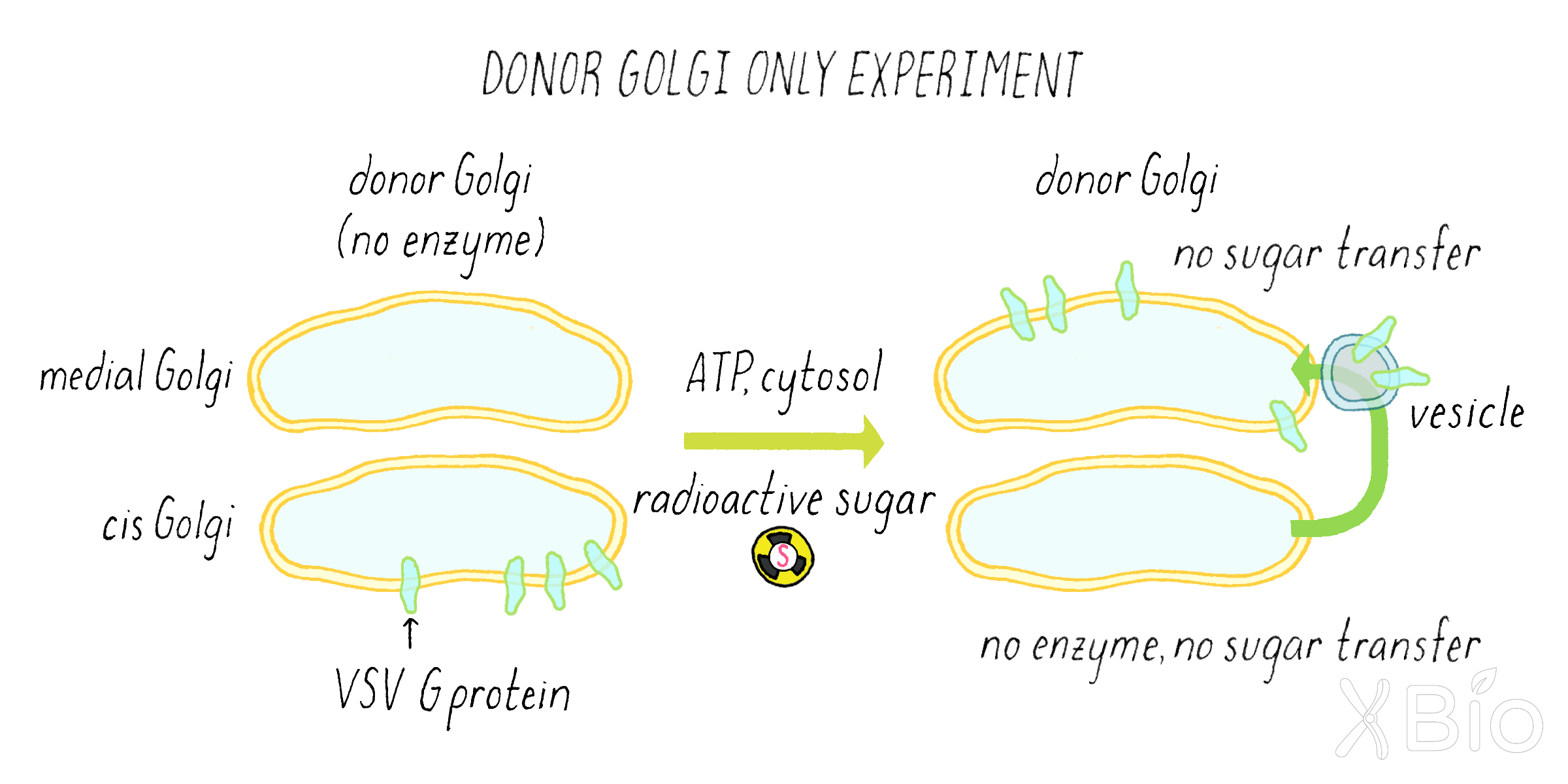
As a strategy for his test-tube reaction, Rothman used a cell line infected with the virus that produces the VSV G protein, but this cell line lacked the enzyme in the medial Golgi that adds the sugar. Thus, in this mutant cell line, when the VSV G protein is transferred from the cis to the medial-Golgi, no sugar is added (Figure 13, right). Rothman prepared Golgi membranes from this VSV-infected cell using a cell fractionation approach (see Clue 2) and called it the "donor Golgi."
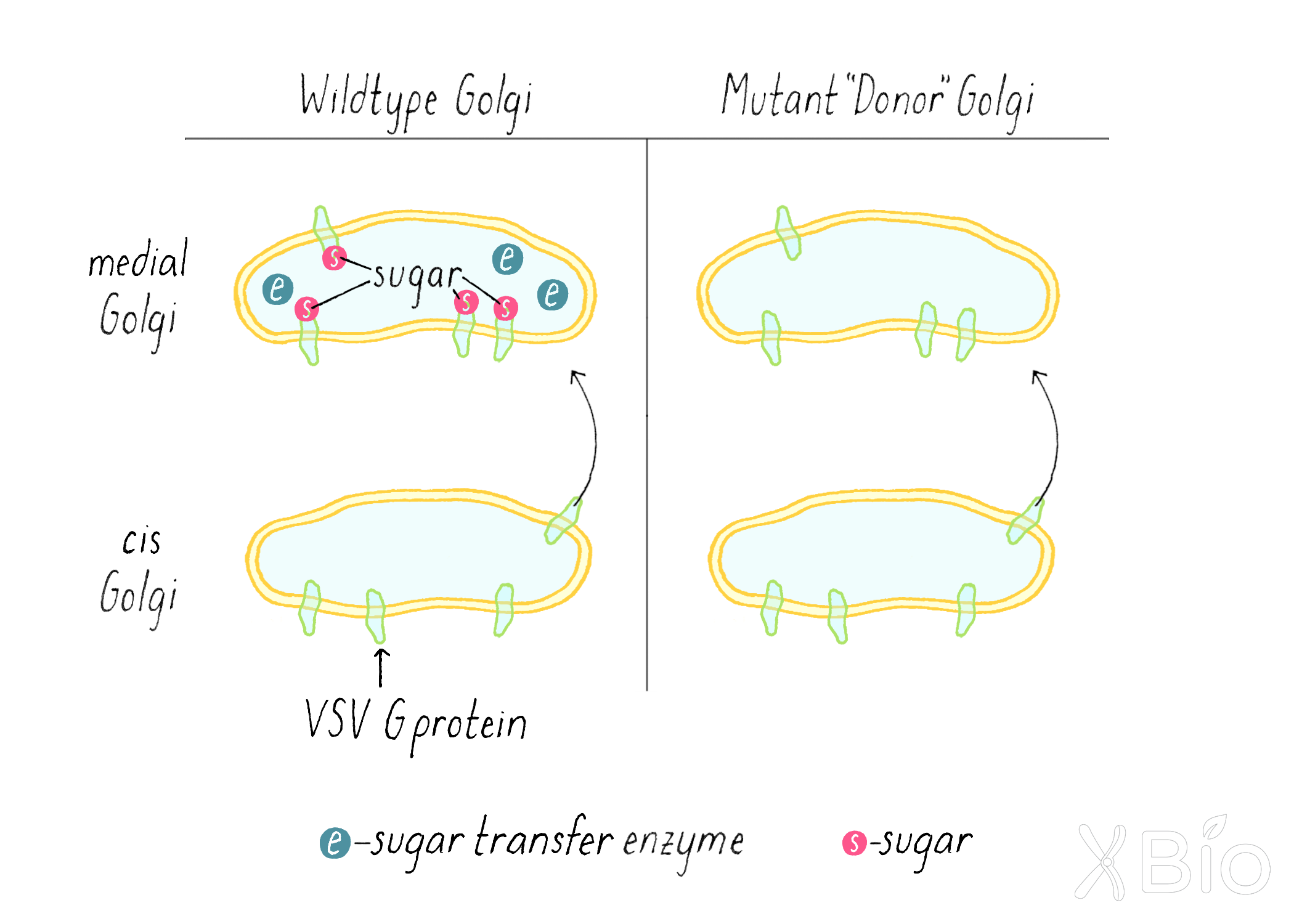
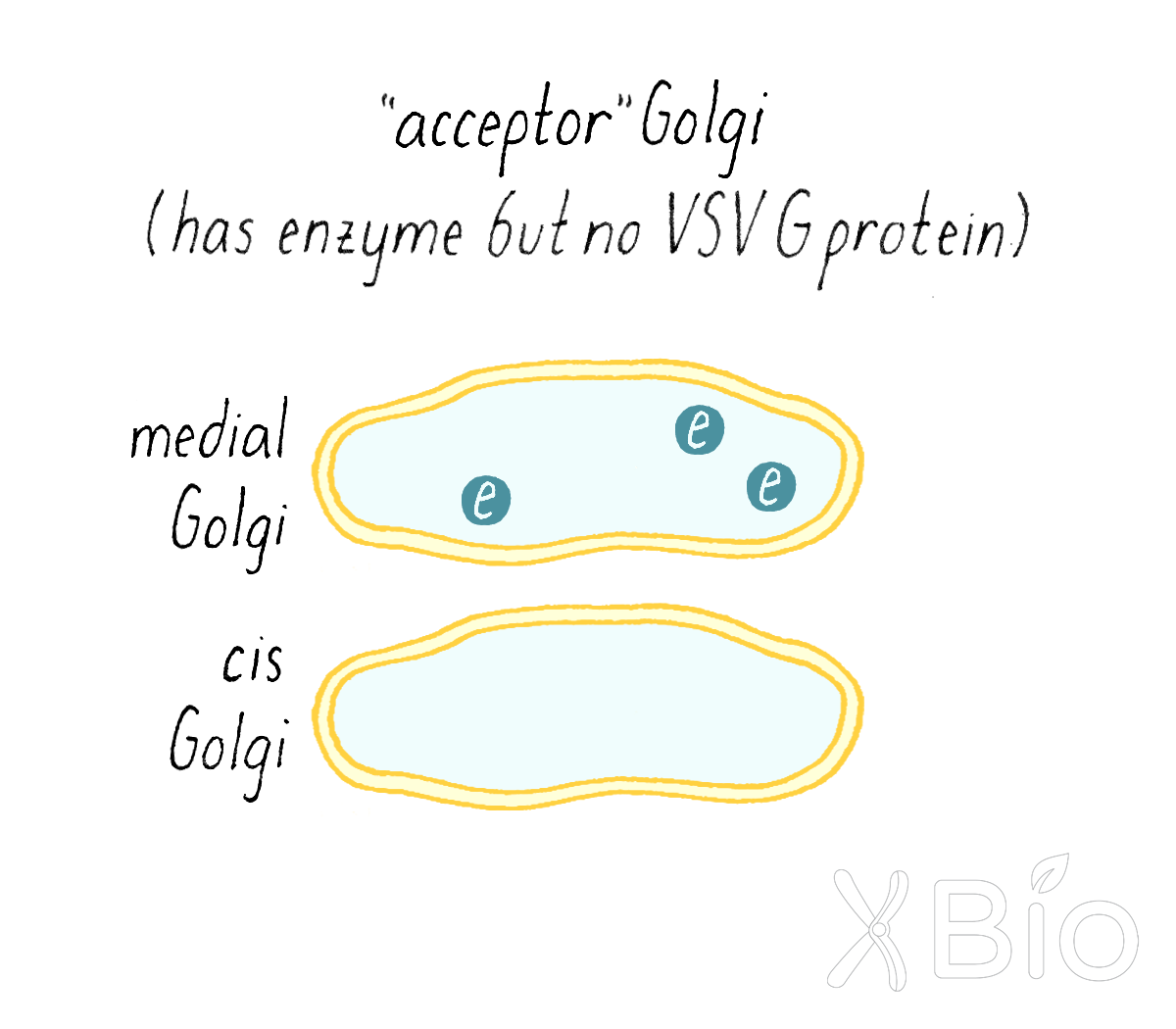
Rothman then prepared a Golgi fraction from a normal mammalian cell line that has the sugar transfer enzyme in the medial Golgi. However, these cells were not infected with virus, so it lacked VSV G protein. This was called the "acceptor Golgi" (Figure 14).
Rothman now combined the acceptor and donor Golgi fractions together in a test tube along with ATP (an energy source for cells), a biochemical cytosol fraction (which contains many free, non-membrane-bound proteins that might be required for the reaction), and the radioactive sugar (Figure 15). He wanted to see if the secretory pathway would still keep working in a test tube. If so, some of the VSV G protein from the donor cis-Golgi of the mutant cells would get transferred to the medial compartment of the normal acceptor Golgi and then get a sugar added to it.
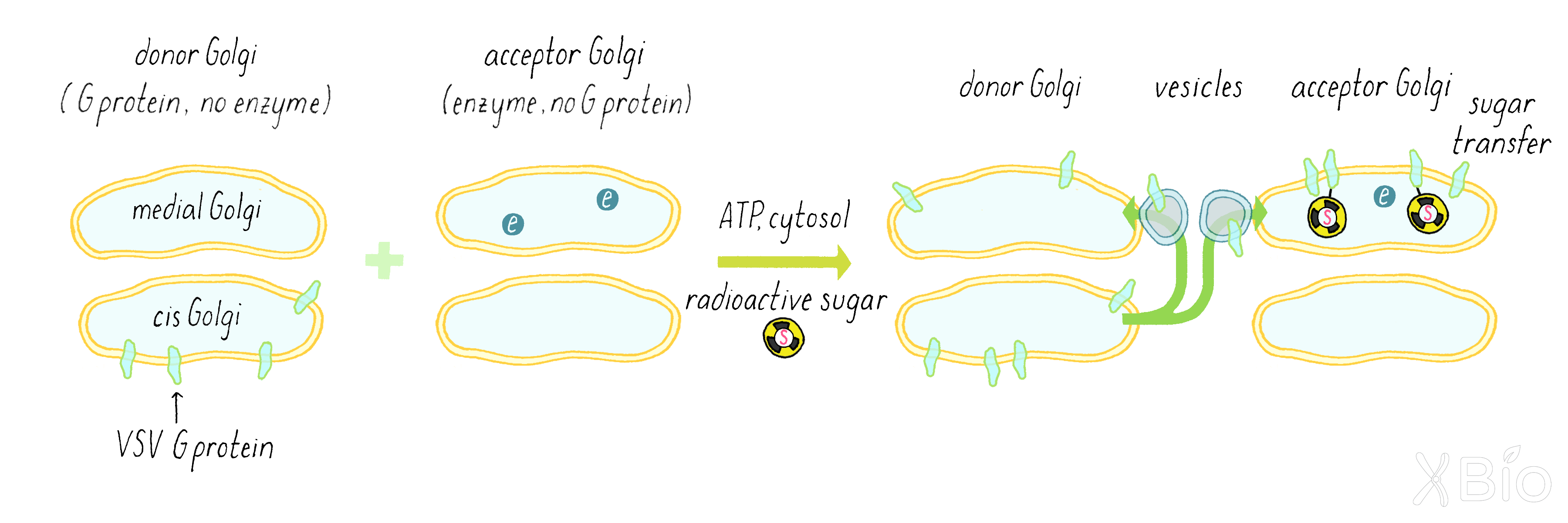
Explorer’s Question: Why did Rothman use a virus-infected cell in this experiment?
Answer: Two reasons. If he wanted to follow the movement from the cis to the medial Golgi, he had to track a unique protein that was not already present in the acceptor medial Golgi compartment. Since donor and acceptor Golgi are from the same type of mammalian cell, virtually all proteins will be the same (except for one sugar enzyme). Second reason is that virus makes a lot of VGV G protein; it is much more abundant than any normal cellular protein. That makes it easier to measure by the technique of immunoprecipitation described below.
By using a radioactive label on the sugar and a technique called immunoprecipitation (see Dig Deeper 2), Rothman measured if radioactive sugar was transferred to the VSV G protein. The biochemical assay clearly revealed the addition of the radioactivity sugar to the VSV-G protein (Figure 16), suggesting that a delivery process from the donor Golgi to the acceptor Golgi continued in the test tube.
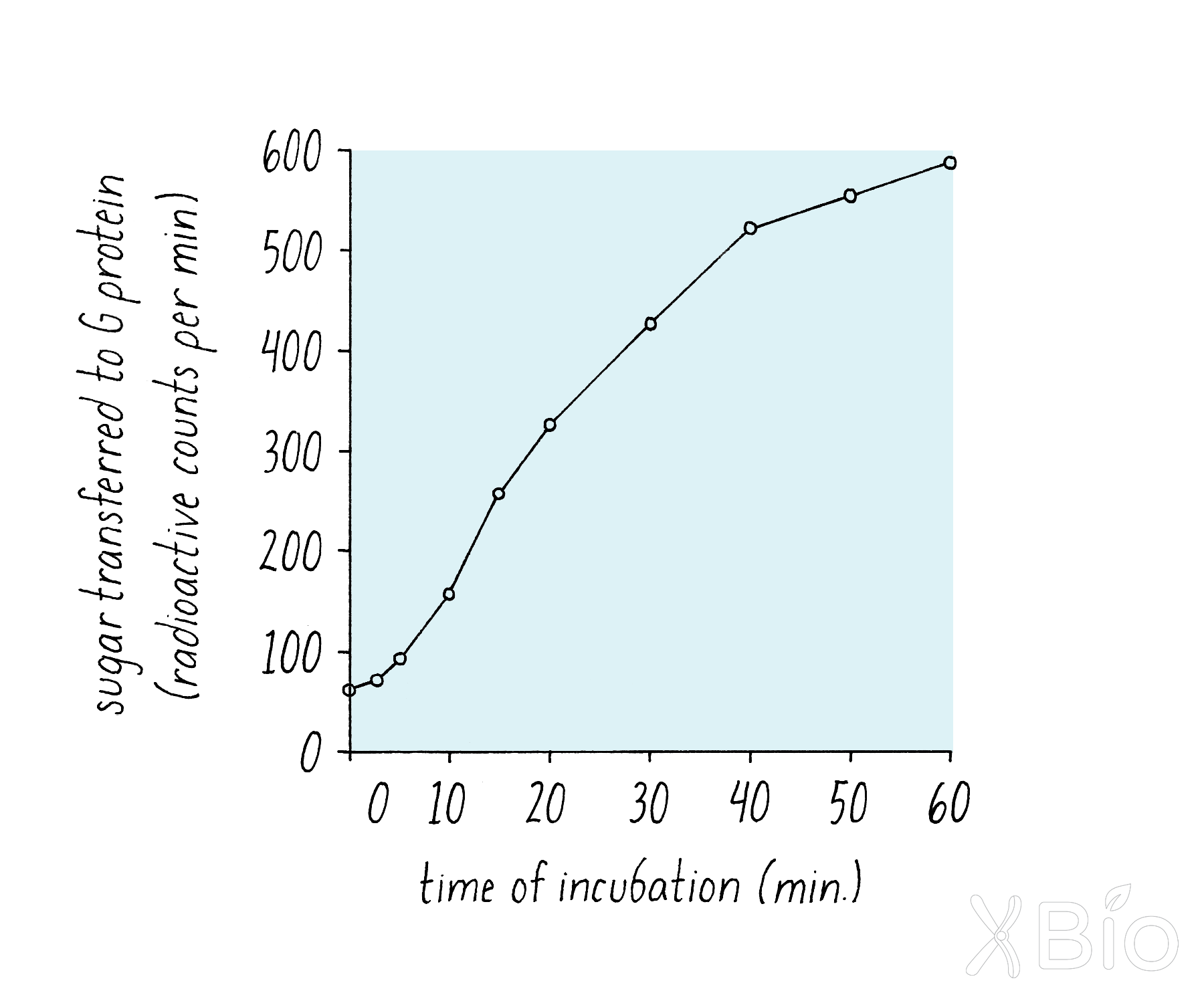
Explorer’s Question: Why did Rothman make different measurements over time as opposed to doing just one time point?
Answer: Doing time measurements makes the data more convincing. If only one time point was measured (e.g., 400 counts), it is possible that it was due to some artefact (e.g., radioactive sugar somehow being scored but not being transferred to the protein). The linear increase over time was consistent with a time-dependent biological reaction, such as might be expected for the transfer of VSV G from the cis- to the medial-Golgi.
Rothman did various additional experiments (called "control experiments") to understand the interpretation of the simple graph in Figure 15. He tested whether the sugar transfer indeed required the enzyme in the acceptor compartment. He did so by leaving out the acceptor compartment entirely from the test tube reaction. In this experiment, he found no sugar transfer (Figure 17). Rothman also did many other experiments to rule out the leakage of enzyme and VSV G proteins, so that he could be assured that the reaction was happening inside of the membranes.
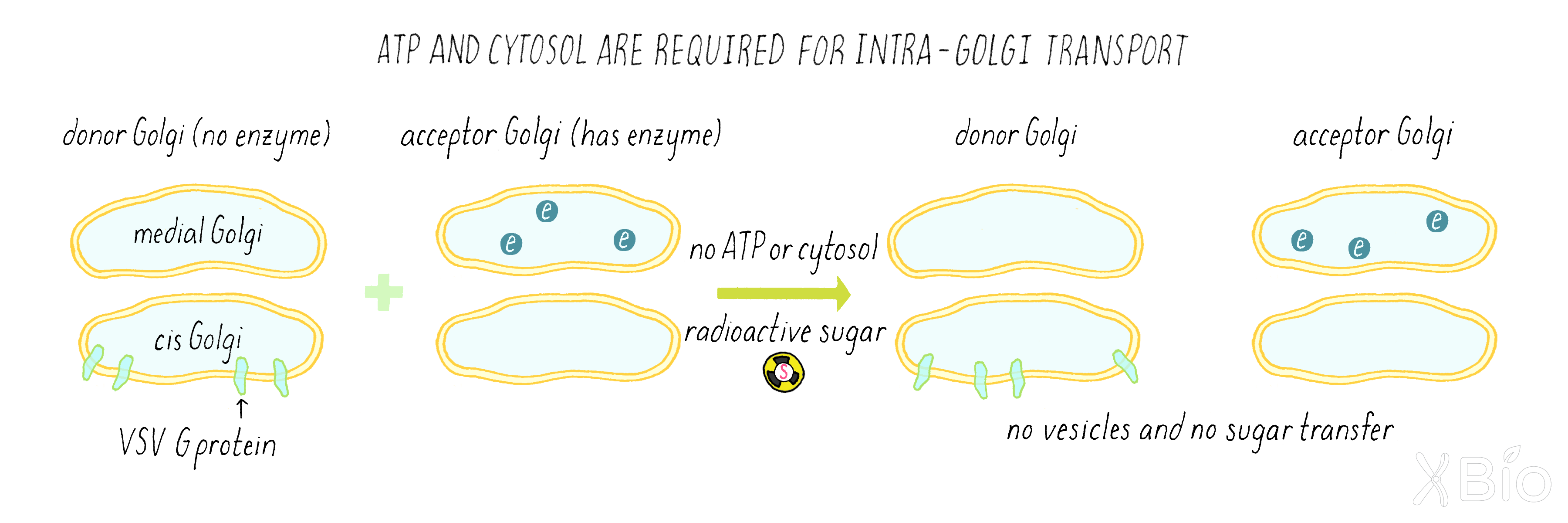
To establish the requirements for the reaction, he left out either ATP or the cytosol protein. In these experiments, there was no sugar transfer either (Figure 18).
Explorer’s Question: What did experiment in Figure 17 reveal about the process of Golgi transport?
Answer: The requirement for ATP indicated that the transfer of proteins between Golgi compartments requires energy. The requirement for cytosol indicated that additional factors (likely proteins) not attached to the membranes themselves are also required.
Rothman discovered several key proteins of the vesicular transport machinery using this in vitro transport assay. These included proteins that promoted budding of vesicles from one membrane compartment and other proteins that allowed fusion of those vesicles with another membrane compartment. We will cover some of these proteins in the Knowledge Overview.
What Happened Next?
Machinery for secretory transport is validated by convergent results from complementary approaches
Remarkably, several years after the discoveries described above, Rothman's results with mammalian cells and Schekman's results with yeast converged. Some of the sec proteins identified in the yeast genetics screen were found to be essentially the same as the proteins identified by Rothman in his mammalian-based biochemical assays. This result revealed that many proteins involved in the secretion pathway are conserved from yeast to mammals.
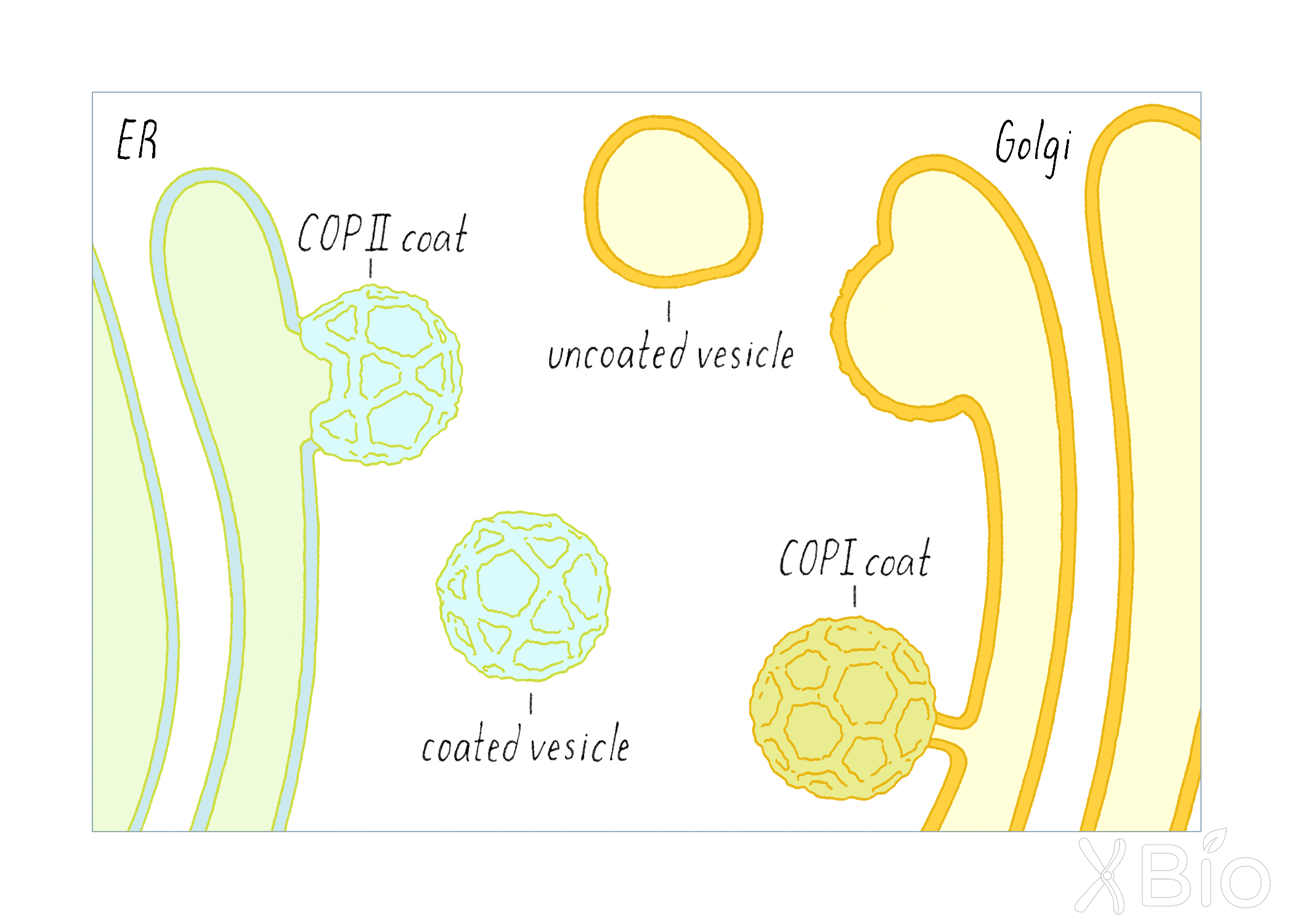
The Schekman and Rothman studies also uncovered proteins that allow the budding of transport vesicles from a membrane compartment. These specialized proteins are called "coat proteins." These proteins form a cage-like structure that helps to facilitate the budding and release of a vesicle (Figure 19).
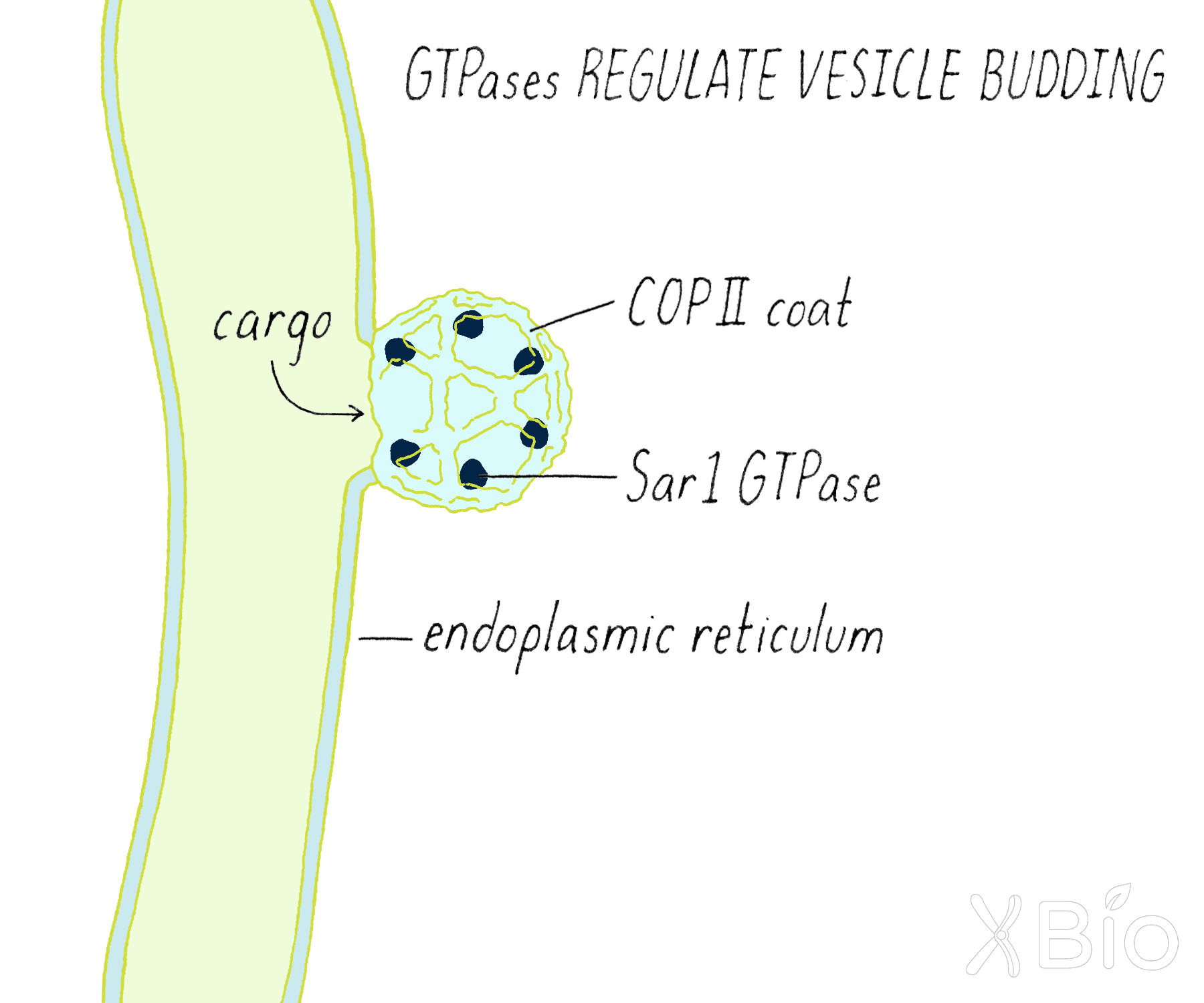
- COPII coats (discovered by Schekman) are involved the formation of vesicles from the ER, which are destined to travel to the Golgi. The locations in the ER where COPII initiates this process are called ER exit sites.
-COPI coats (discovered by Rothman) help form vesicles at the Golgi, which regulate recycling of materials back to the ER.
After budding, the vesicles release their coats as they journey between the ER and the Golgi.
Two very general lessons emerged from these results:
1) Genetics and biochemical approaches are both powerful and complementary.
2) The similar findings of Schekman and Rothman highlight the fact that many fundamental processes are conserved across all eukaryotes, from baker's yeast to humans. Studies in model organisms thus can be relevant to human biology and provide insight into diseases.
Live cell imaging reveals the dynamics of secretory traffic, including the role of retrograde transport
While Schekman and Rothman used genetics and biochemistry to study protein secretion, other groups, including my own lab, started using light microscopy to follow the movement of secretory proteins through the secretory pathway. We decided to track VSV G, the same protein used by Rothman, by tagging it with the green fluorescent protein (GFP; see Key Experiment by Chalfie). We wanted to synchronize the traffic of the VSV G, so that the protein's transport started in the ER and then moved at the same time through the secretory process. This was analogous to the Palade and Jameson experiment described in Clue 2; however, instead of looking at fixed cells at different time points, we examined a single living cell by fluorescence microscopy over time, tracking the movement of cargo continuously through the pathway. To do so, we used a trick that was already introduced in the Schekman experiments—a temperature-sensitive (ts) mutation. Previously, a VSV G protein with a mutation was identified that caused it to be trapped within the ER at the restrictive temperature of 40°C, but permitted normal movement through the secretory pathway at 32°C. In our experiment, we trapped the mutant GFP-tagged VSV G in the ER at 40°C, shifted the temperature suddenly to 32°C, and then followed the synchronous wave of movement as the protein left the ER and proceeded through the secretory pathway (Figure 20).
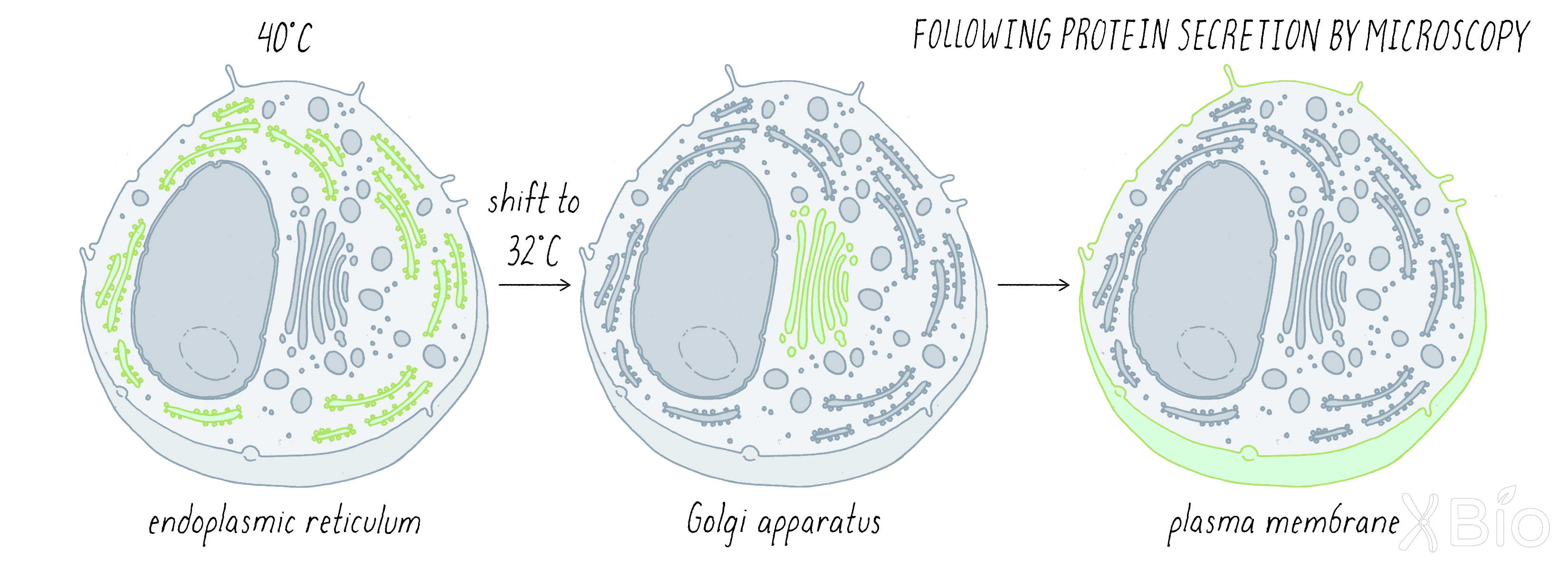
Our live cell imaging experiments revealed that GFP-tagged VSV G protein emerged from the ER at specialized sites called ER exit sites scattered across the elaborate ER network. The ER exit sites are enriched in the budding factors identified by Schekman and Rothman, including COP II and COP I, consistent with their role in secretory trafficking. Vesicles loaded with VSV G bud from these ER exit sites and move toward the Golgi apparatus along microtubule tracks (Video 3). Upon reaching the Golgi, the transport intermediates fused with the Golgi, releasing VSV G into the Golgi system. After spending a short time in the Golgi, VSV G is then packaged into a different set of transport vesicles and tubules that bud out of the Golgi and travel to the plasma membrane.
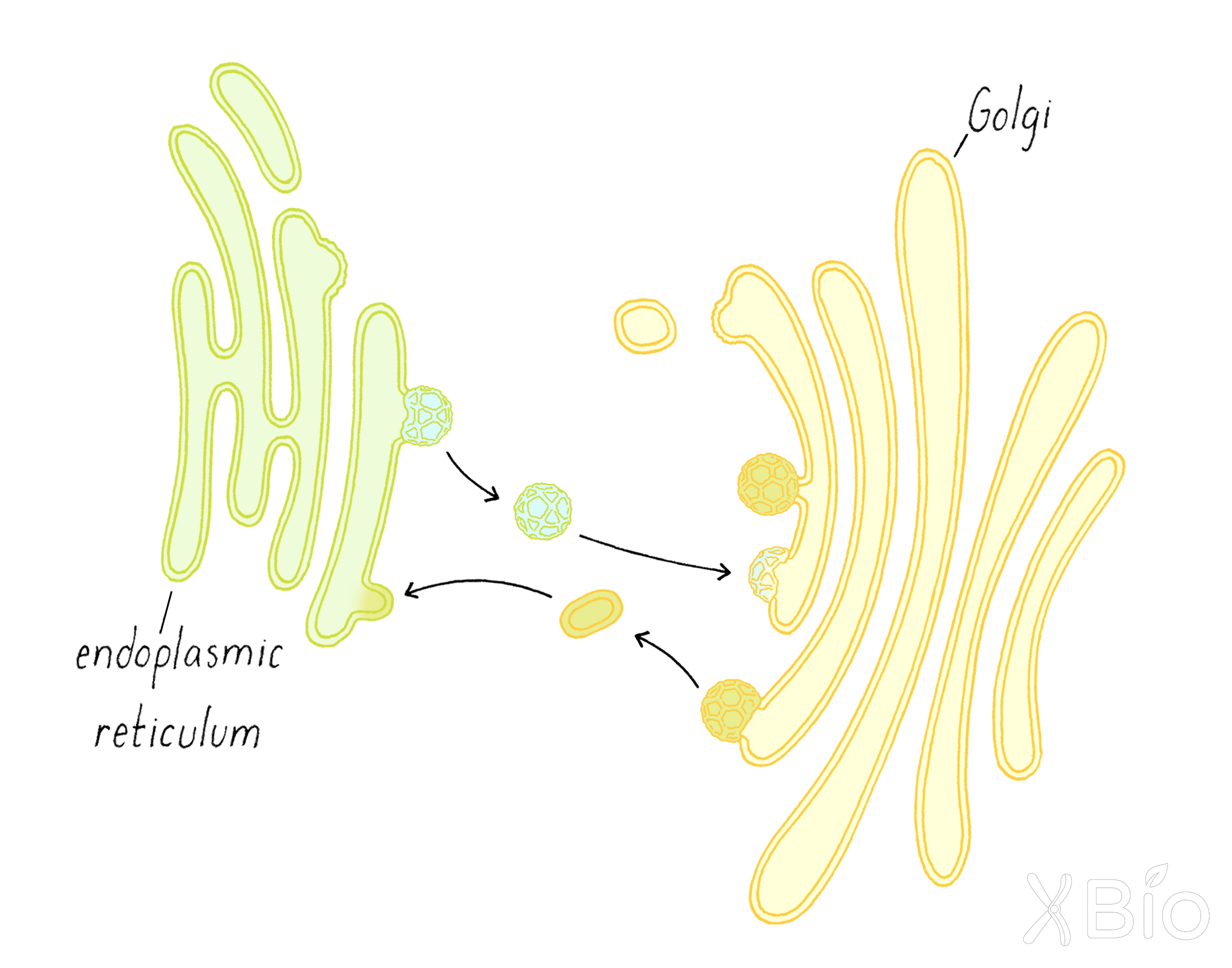
The model of the secretory pathway, as presented in Figure 8 and visualized in Video 3, suggests a one-way transport system through "permanent" organelles such as the Golgi. However, a different view of membrane trafficking emerged serendipitously from imaging cells treated with a compound (brefeldin A [BFA]) that inhibits secretion. Within minutes of treating with BFA, the entire Golgi apparatus blew apart into tubules and the tubules carried Golgi proteins back into the ER (Video 4). When BFA was removed from cells, we found that a fully functional Golgi re-emerged within minutes (Video 4). Subsequent studies delving into the molecular basis of BFA action revealed it inactivates a protein that acts to recruit COPI to membranes.
Overall, these studies showed two-way traffic between the ER and Golgi, not just one way as previously thought. As we learned from the Palade experiment in Clue 2, the ER is continually shipping material to the Golgi. However, some proteins that leave the ER are part of the delivery system to the Golgi but are not destined for secretion. These delivery proteins need to be recycled from the Golgi back to the ER so that they can be used again. In the presence of BFA, the forward transport from ER to Golgi is blocked, while the reverse transport from Golgi to ER continues. As a result, the Golgi "disappears" back into the ER. These results suggested that the Golgi undergoes continuous outgrowth from and re-consumption by the ER through bidirectional membrane trafficking (Figure 21).
To sum up, while biochemical and genetic approaches provided dramatic advances in understanding the secretory pathway, the development of new imaging technologies allowed the field to better understand the dynamics of the secretory pathway. Another technique called fluorescence recovery after photobleaching (FRAP) (Dig Deeper 3) also allowed us to show that proteins are moving about rapidly within a membrane compartment, as well as between them. We will learn more about the organelles of the cell and the proteins that are involved in membrane trafficking in the Knowledge Overview.
Part II: Knowledge Overview —
The Internal Membrane Systems of Eukaryotic Cells
Membrane-enclosed compartments, called organelles, are a ubiquitous feature of eukaryotic cells, occupying approximately one-third of cytoplasmic volume in a typical cell. Composed of an internal lumen with a unique chemical composition and a surrounding semipermeable membrane, organelles are specialized for particular functions that are essential for proper cell behavior and metabolism. These include roles in energy production and in the synthesis/storage/processing/degradation of macromolecules. Compartmentalizing these functions within organelles allows cells to more efficiently carry out complex biochemical reactions and to isolate potentially harmful chemical products from the rest of the cell.
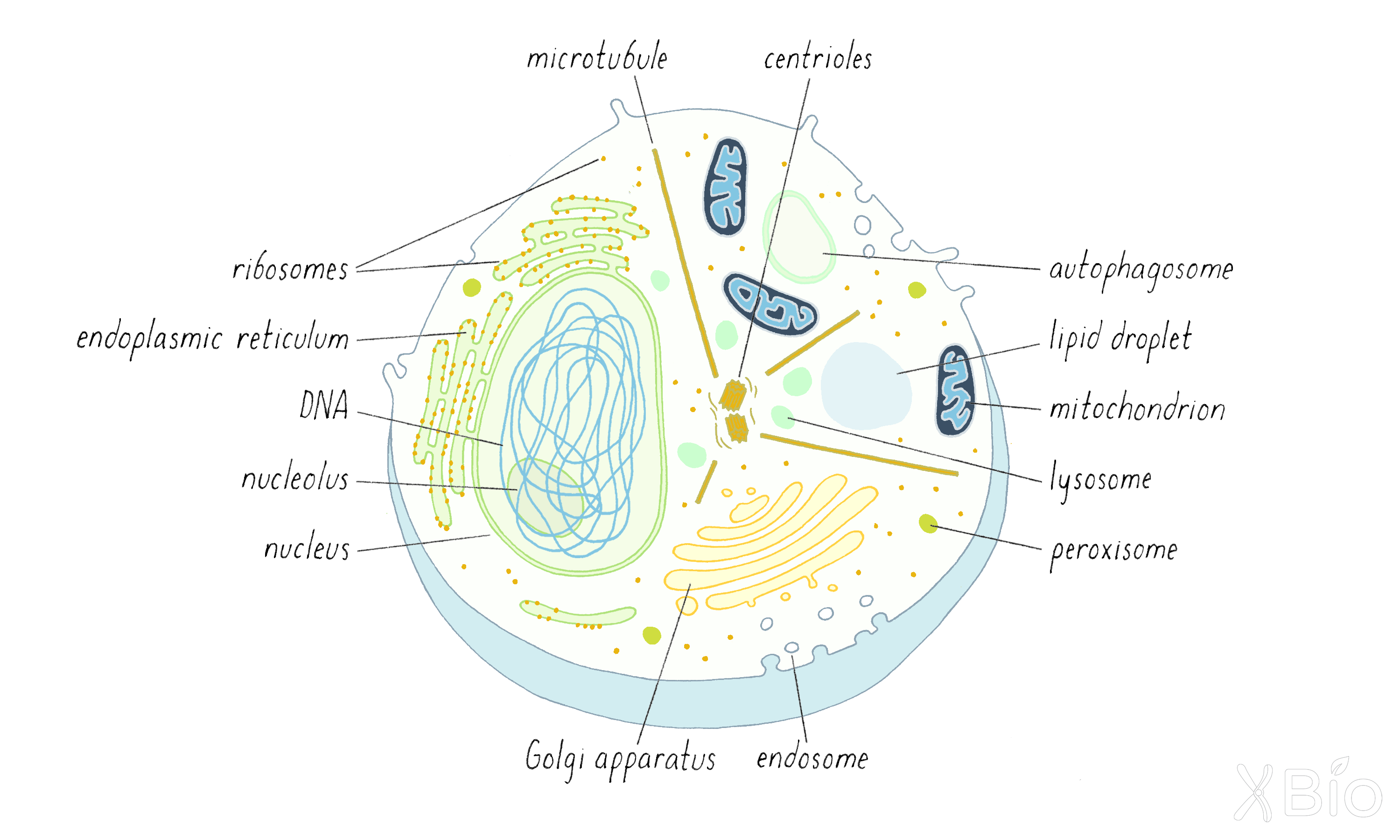
There are nine distinct membrane-bound organelles in all eukaryotic cells (Figure 22):
1) nucleus,
2) endoplasmic reticulum (ER),
3) Golgi apparatus,
4) endosome,
5) autophagosome,
6) lysosome,
7) mitochondria,
8) lipid droplet, and
9) peroxisome
A movie (Video 5) of several organelles labeled with different colored fluorescent proteins also shows that they have very different shapes and occupy distinct positions within the same cell.
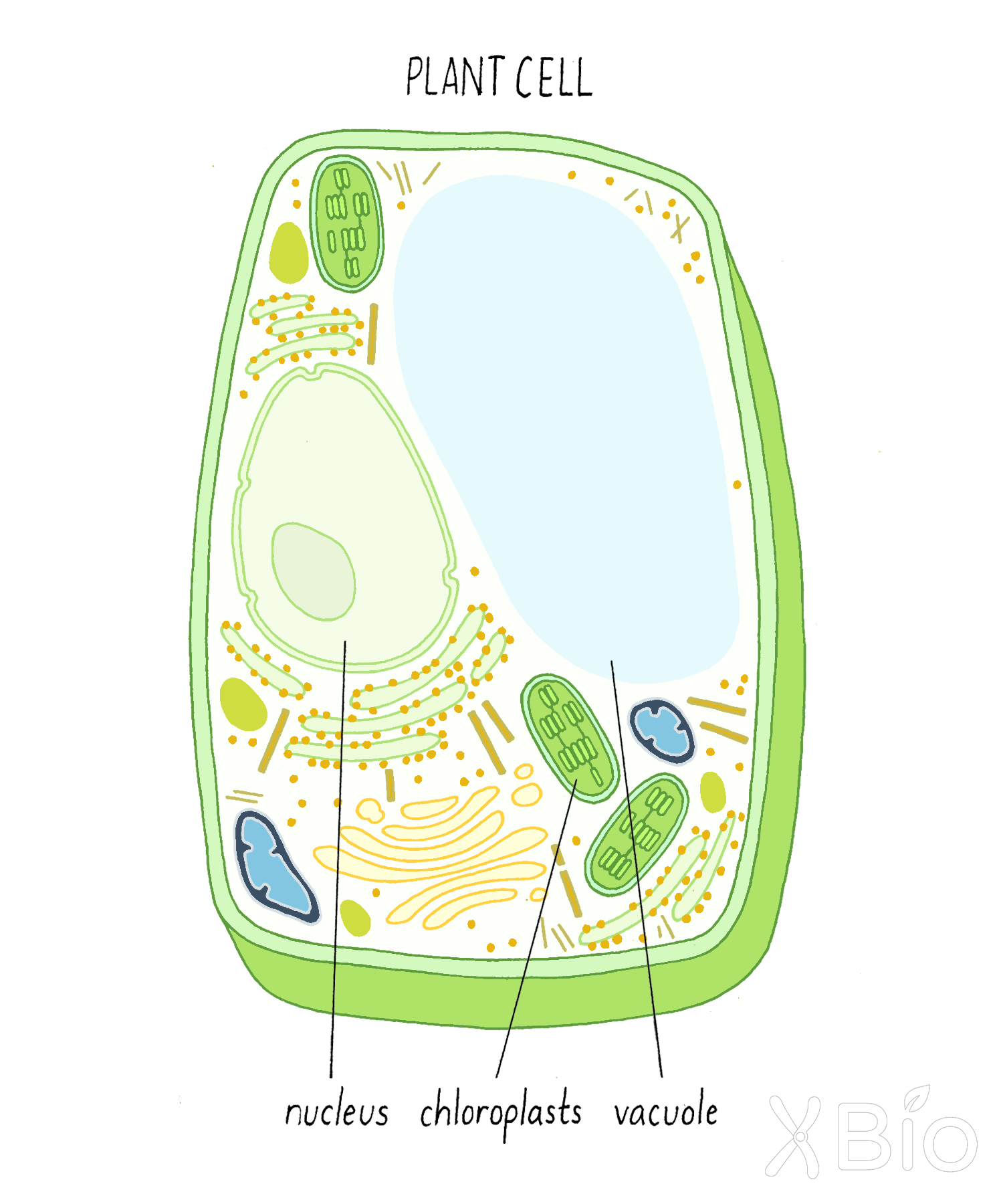
In addition, plants and algae have chloroplasts and vacuoles (Figure 23). Chloroplasts convert light (e.g., from the sun) into energy through photosynthesis. Vacuoles, a variation of a lysosome, have degradative functions like lysosomes but also can fill with water and ions and maintain pressure against the cell wall (turgor pressure).
30% of the different proteins in eukaryotic cells are found either in the membranes or within the lumens of organelles. Simple bacteria and archaeal cells, which lack internal membrane-bound compartments and the genes coding for organelle-associated proteins, do not have the morphological diversity and adaptability of eukaryotic cells, which comprise all multicellular life forms on earth.
Eukaryotic organelles perform four major functions:
1. Take up and digest macromolecules from outside the cell. Endocytosis (endosomes) take up extracellular molecules. Autophagosomes take up intracellular material. Lysosomes mediate the final digestion for both.
2. Synthesize, process, and transport proteins. This occurs through the activities of the ER, Golgi apparatus, and their transport intermediates, which form the secretory pathway.
3. Harness or direct energy-producing pathways, as well as store and oxidize lipid molecules. This is the job of mitochondria, lipid droplets, and peroxisomes.
4. House and control the cell's genetic material, which is performed by the nucleus.
What is a membrane?
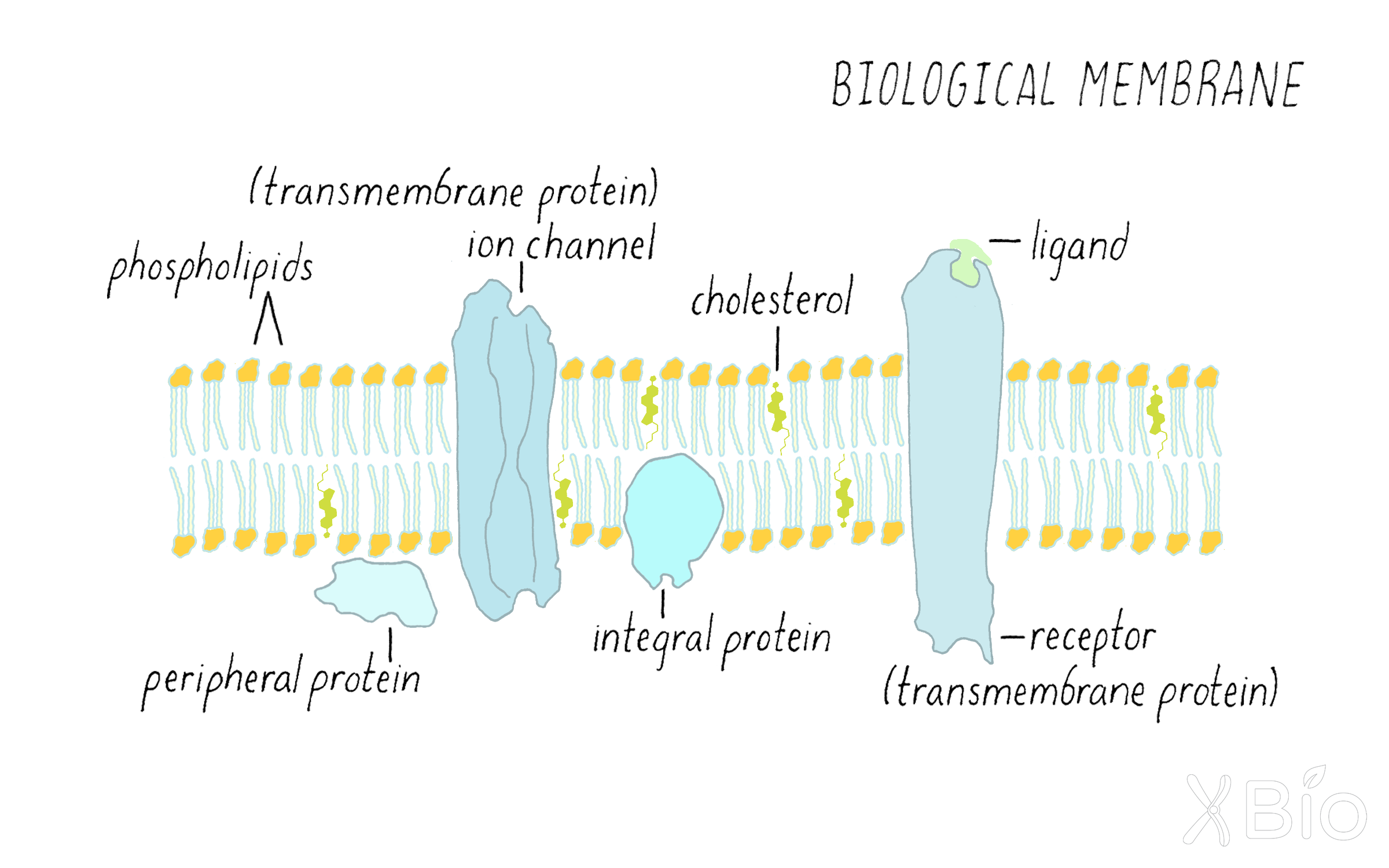
To help understand how these organelles differentiate themselves and function in a cellular context, it is worth considering the properties of their enclosing membranes. Eukaryotic cell membranes are composed of fluid lipid bilayers with embedded and peripherally associated proteins that are highly dynamic (Figure 24). We know this from several pieces of evidence. First, membranes split into two while frozen (a technique called freeze fracturing) and then viewed by electron microscopy reveals two lipid leaflets and the presence of protein particles embedded in the bilayer. Second, chemical labeling approaches show a single protein can be exposed to the two different sides of the membrane. Third, live cell light microscopy and other approaches further reveal that lipids and proteins can undergo free lateral motion in the plane of the membrane, and that lipids can flip from one side of the membrane to the other.
Lipids
Biological membranes are composed of lipids. Lipids consist mostly of water-repelling (hydrophobic) hydrocarbon chains attached to a water-loving (hydrophilic) polar headgroup (Figure 25). This special character allows lipids to spontaneously self-assemble into ordered bilayer structures when placed in an aqueous environment. Within such a bilayer, the lipid's hydrocarbon chains face inside and polar groups face the surrounding water. Proteins residing within the lipid membrane bilayers float freely unless restricted through interactions with themselves or with networks of cytoplasmic proteins.
Three major classes of lipids are found in cell membranes of all eukaryotes (Figure 24)—glycerophospholipids, sphingolipids, and sterols.
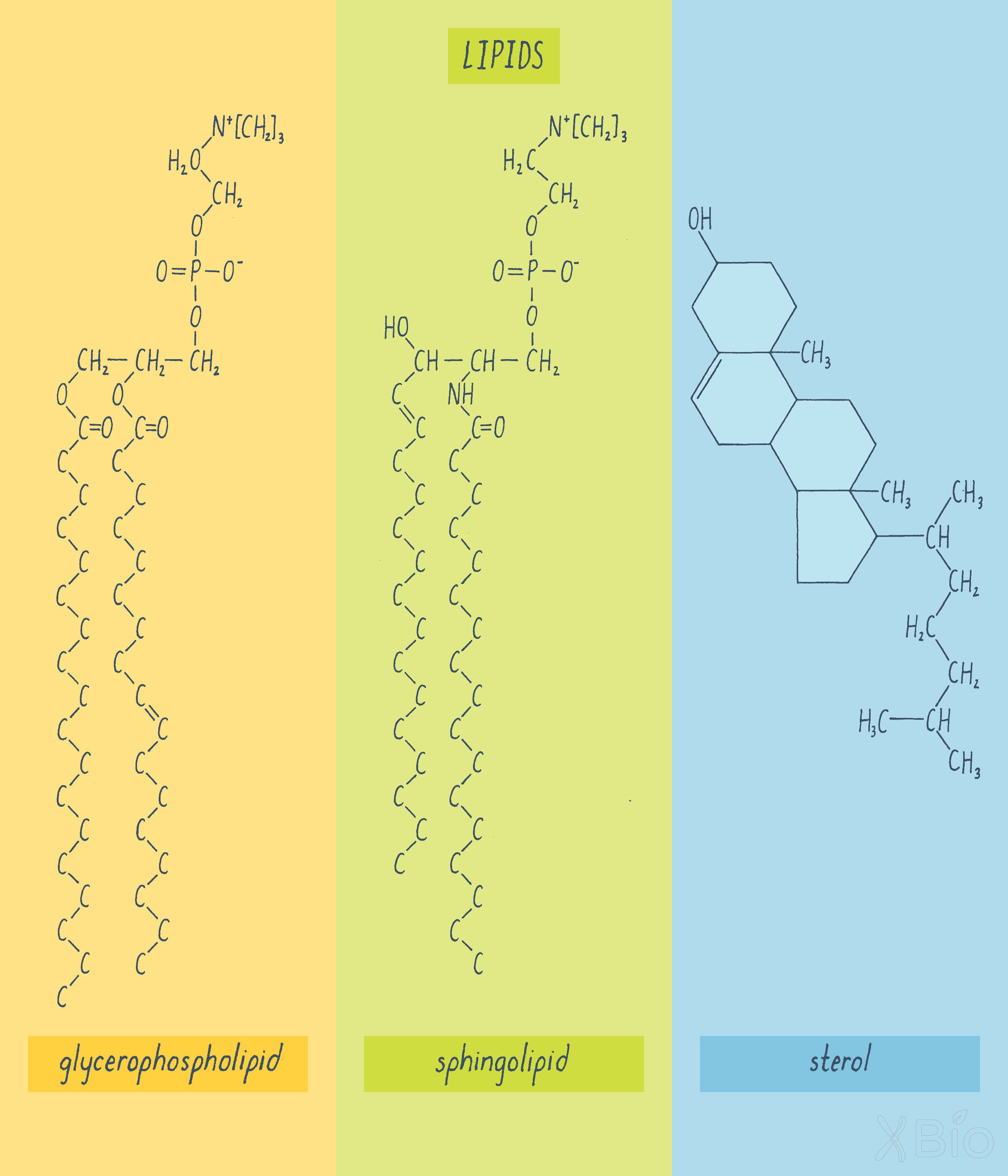
Glycerophospholipids
Glycerophospholipids are the most abundant class and consist of a three-carbon backbone of glycerol, two long-chain fatty acids esterified to the glycerol, and a phosphoric acid esterified to the remaining carbon of the glycerol. Certain proteins, called phospholipid translocators, flip negatively charged phospholipids like phosphatidylserine to the cytosolic-facing leaflet of the bilayer, which keeps the compositions of the inner and outer leaflets of the bilayer distinct.
A protein can move within the plane of the membrane through the random process of diffusion. How fast the protein moves depends upon the viscosity of the membrane or also referred to as its fluidity. As the viscosity of the membrane increases (e.g., from liquid-like to more molasses-like), the membrane becomes less fluid, resulting in proteins diffusing more slowly in the membrane. Double bonds in the fatty acid chain of glycerophospholipids, which create a bend along the fatty acid chain's length, increase membrane fluidity. Addition of more sphingolipids and sterols (discussed below) makes membranes less fluid.
Sphingolipids
Sphingolipids are a chemically distinct group of lipids generally found on the outer leaflet of the plasma membrane. They decrease membrane fluidity and also make the membrane more resistant to mechanical or chemical perturbation.
Sterols
Sterols, the third class of membrane lipids, include cholesterol, which makes up approximately a quarter of the lipids in the plasma membrane. Sterols, which consist of a rigid four-ring structure, resides within the core of the lipid bilayer and can interact with and stabilizing nearby glycerophospholipids and sphingolipids. Cholesterol is synthesized directly by the cell, but it can also be taken up from the diet. Most ingested cholesterol (i.e., from egg yolks, meat, and cheese), however, is only slowly absorbed by the body. Cholesterol is essential for life, but too much of it in your blood can result in the formation of plaques in coronary arteries, which gives rise to the disease called atherosclerosis.
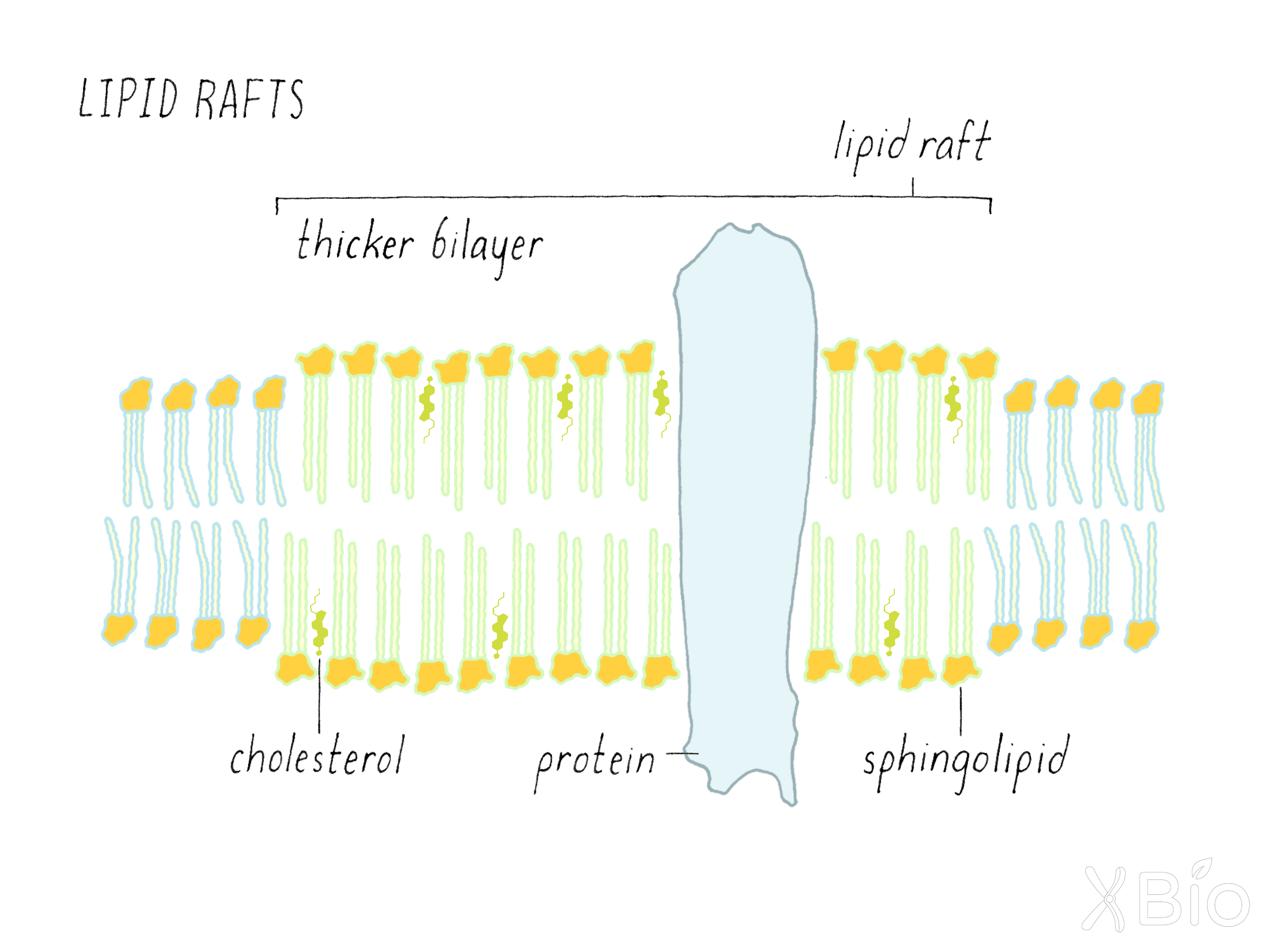
Interactions among the three classes of lipids (i.e., glycerophospholipids, sphingolipids, and sterols) enable eukaryotic cells to generate biophysical and biochemical differences in their membranes, leading to the formation of subdomains. A lipid raft is an example of a subdomain within the plasma membranes (Figure 26). Lipid rafts form when sphingolipids become concentrated with cholesterol, due to cholesterol's preferential interactions with the long, straight acyl chains of sphingolipids. The resulting lipid subdomains are thicker, less fluid, and less permeable to water molecules than is a bilayer consisting of glycerophospholipids alone. Their thicker diameter also allows them to attract and retain certain membrane proteins with longer hydrophobic transmembrane domains. The functions of lipid rafts are still being debated, but they are thought to serve specific roles in cell signaling and the formation of transport vesicles.
Overview of the nine conserved eukaryotic organelles
An overview of the cell's organelles is provided in Video 6.
Let us next survey the roles of the nine conserved eukaryotic organelles. An easy way to remember and group organelles is based on their overall functions, with one group involved in storage/biosynthesis/processing of molecules (i.e., nucleus, ER, Golgi), another group involved in the uptake, degradation/detoxification of substrates (i.e., endosomes, lysosomes, autophagosomes, and peroxisomes), and a final one involved in energy production (i.e., mitochondria).
Nucleus
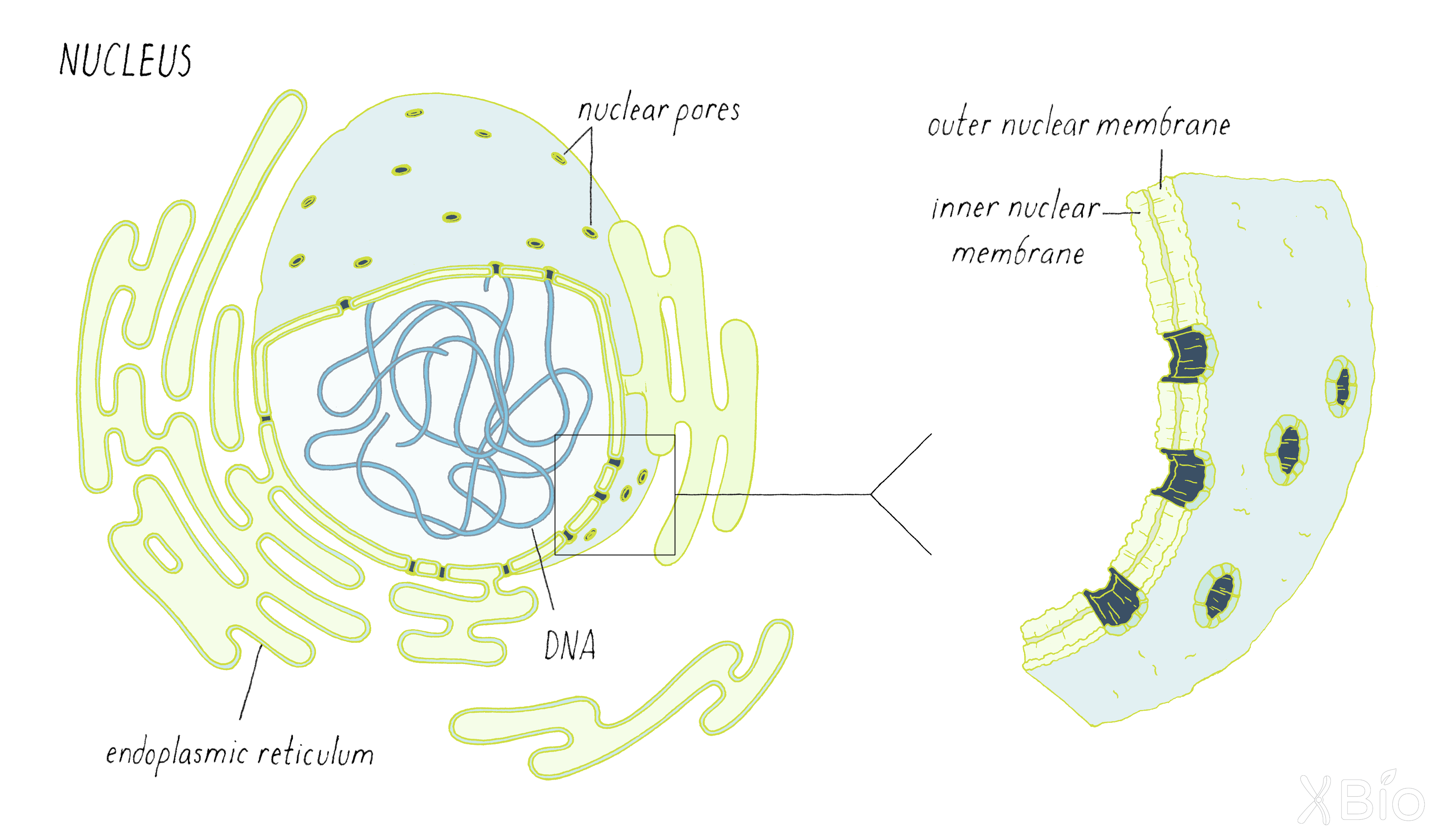
The nucleus (Figure 27) contains the cell's genetic material, which is composed of DNA organized into chromosomes. It is surrounded by two membrane bilayers, the inner and outer nuclear membrane, and the outer membrane is continuous with the ER. The nucleus is studded with nuclear pores that allow proteins and mRNA to move between the nucleus and cytoplasm. As discussed below, separating nuclear activities from cytoplasmic activities through the formation of the nucleus and its envelope was likely an early event in the evolution of eukaryotic cells.
Endoplasmic reticulum
The ER is the most expansive organelle, extending as a tubular network from the nuclear envelope to the cell periphery. The ER spreads throughout the cell as a spider-like tubular network through the help of molecular motor proteins that pull ER tubules along microtubule tracks (Video 7).
Explorer’s Question: In Video 7, the ER jiggles a lot whereas the microtubules more static along their length. What does this tell you about the two structures?
Answer: This mobility reflects the flexural rigidity of the two structures. A microtubule a cytoskeletal polymer made up of the protein tubulin, has a comparable rigidity to a stiff plastic, such as Plexiglass. The ER jiggles because it is much less stiff and highly flexible.
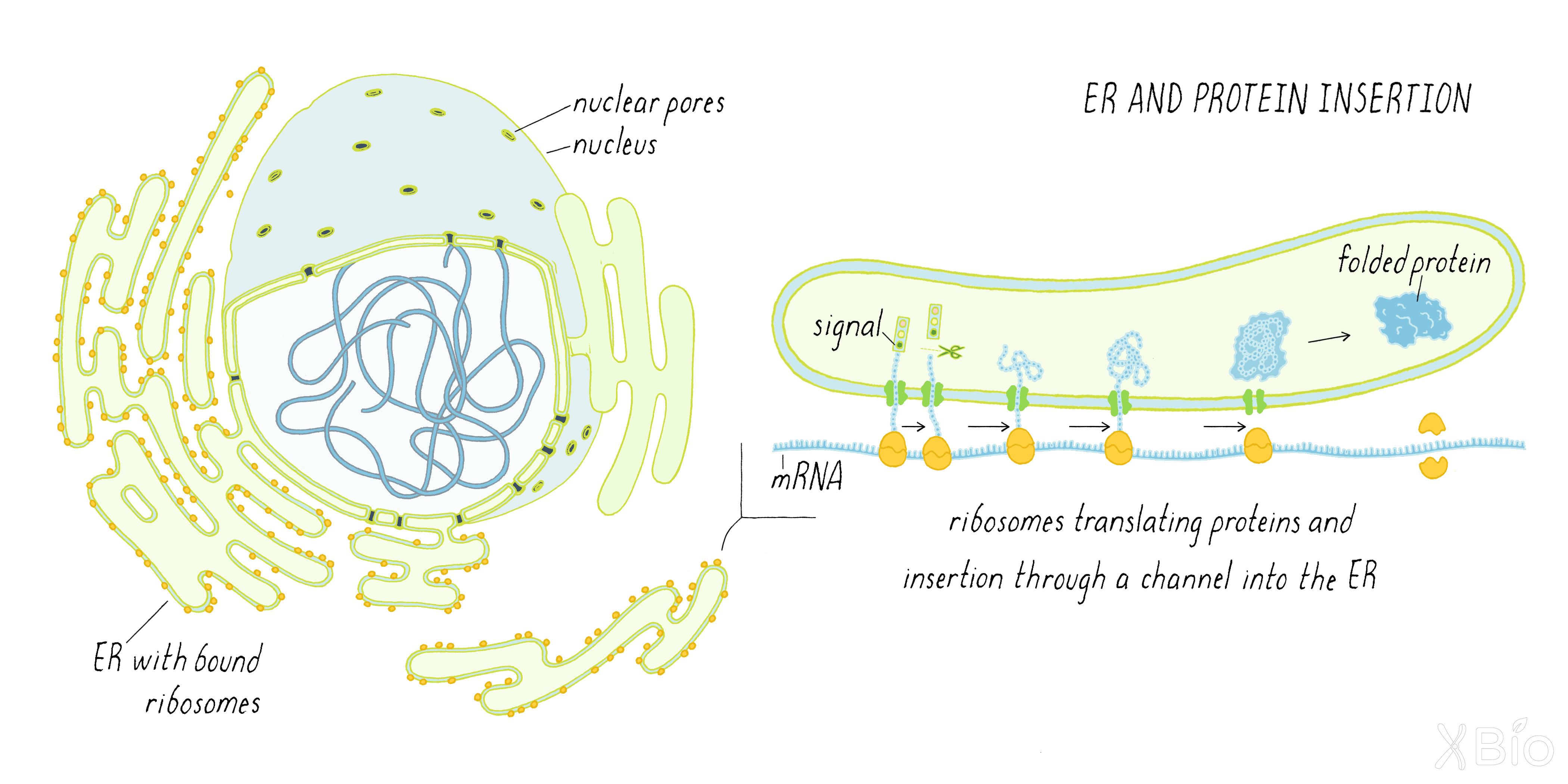
The ER's primary function is to produce the proteins secreted from cells as well as to synthesize the lipids that make up the membranes of all other internal organelles and the plasma membrane. Newly synthesized membrane proteins become inserted into or across the ER's lipid bilayer in areas that are studded with ribosomes (called the rough ER; Figure 28). Specialized receptors and channels on the rough ER's surface drive the transfer of proteins synthesized by ribosomes in the cytoplasm across the ER membrane. Once inside the ER lumen, newly synthesized proteins are exposed to proteins called chaperones that help them fold into their final, active state. Proteins that do not fold correctly in the ER are destroyed by a pathway called ERAD (i.e., ER-associated degradation). Many soluble and membrane proteins that are synthesized in the ER are exported from the ER at sites called ER exit sites. Formation of these sites requires the COPII and COPI coat proteins described by Schekman and Rothman (see Journey to Discovery).
The ER membrane contains calcium pumps that pump Ca2+ into the ER lumen. Ca2+ becomes concentrated in the ER by binding to various ER-localized Ca2+ binding proteins. Signaling pathways can later trigger release of this Ca2+ from the ER to modulate various Ca2+-dependent cellular processes. An example is muscle contraction, which is triggered by release of Ca2+ from the ER.
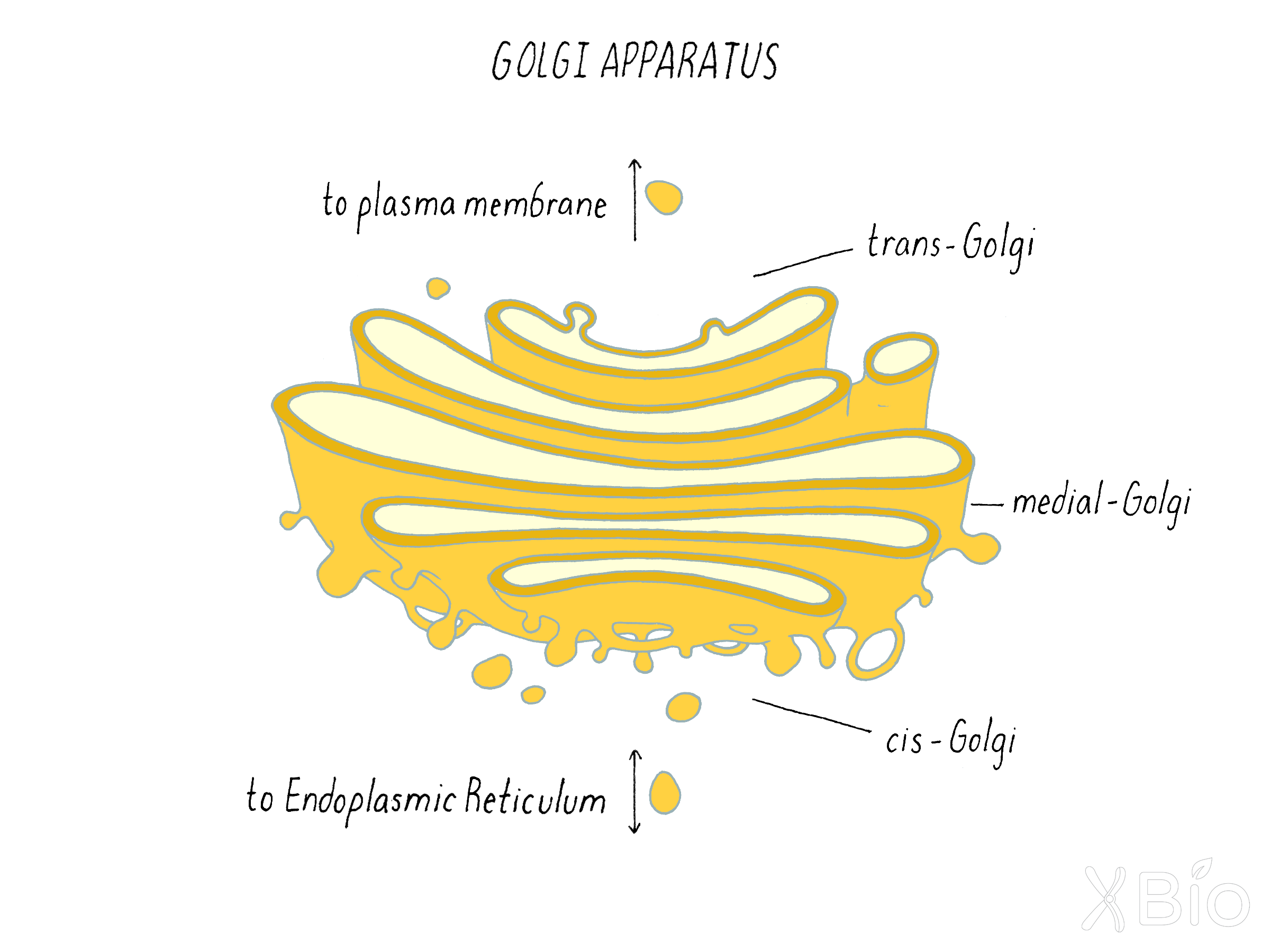
Golgi apparatus
The Golgi apparatus (Figure 29) is composed of a series of flattened, interconnecting cisternae arranged as a stack. Cargo (proteins and lipids) arrive at one face of the stack (the cis-Golgi and leave at the other end (the trans-Golgi). The passage of proteins between Golgi stacks is discussed in the Journey to Discovery, which features experiments by James Rothman.
The Golgi apparatus houses hundreds of enzymes that modify the proteins and lipids that pass through it. An example is glycosylation—a process that covalently attaches sugars to proteins. Proteins that are excreted from the cell or are exposed to the exterior are frequently heavily modified with sugars. Glycosylation begins in the ER, where it plays a critical role in protein assembly and stability, and then continues with many modifications in the Golgi. These later sugar modifications are critical for proteins to function properly outside the cell. Indeed, secreted hormones only properly bind to and activate receptors on neighboring cells if they are correctly glycosylated. Glycosylation in the Golgi apparatus is thus an important and highly regulated mechanism for protein processing within cells.
After modifying molecules derived from ER, the Golgi sorts them into pathways that takes them to either the plasma membrane, lysosomes, endosomes, or back to the ER. Sphingolipids generated in the Golgi help in this sorting process by creating raft domains that facilitate Golgi export.
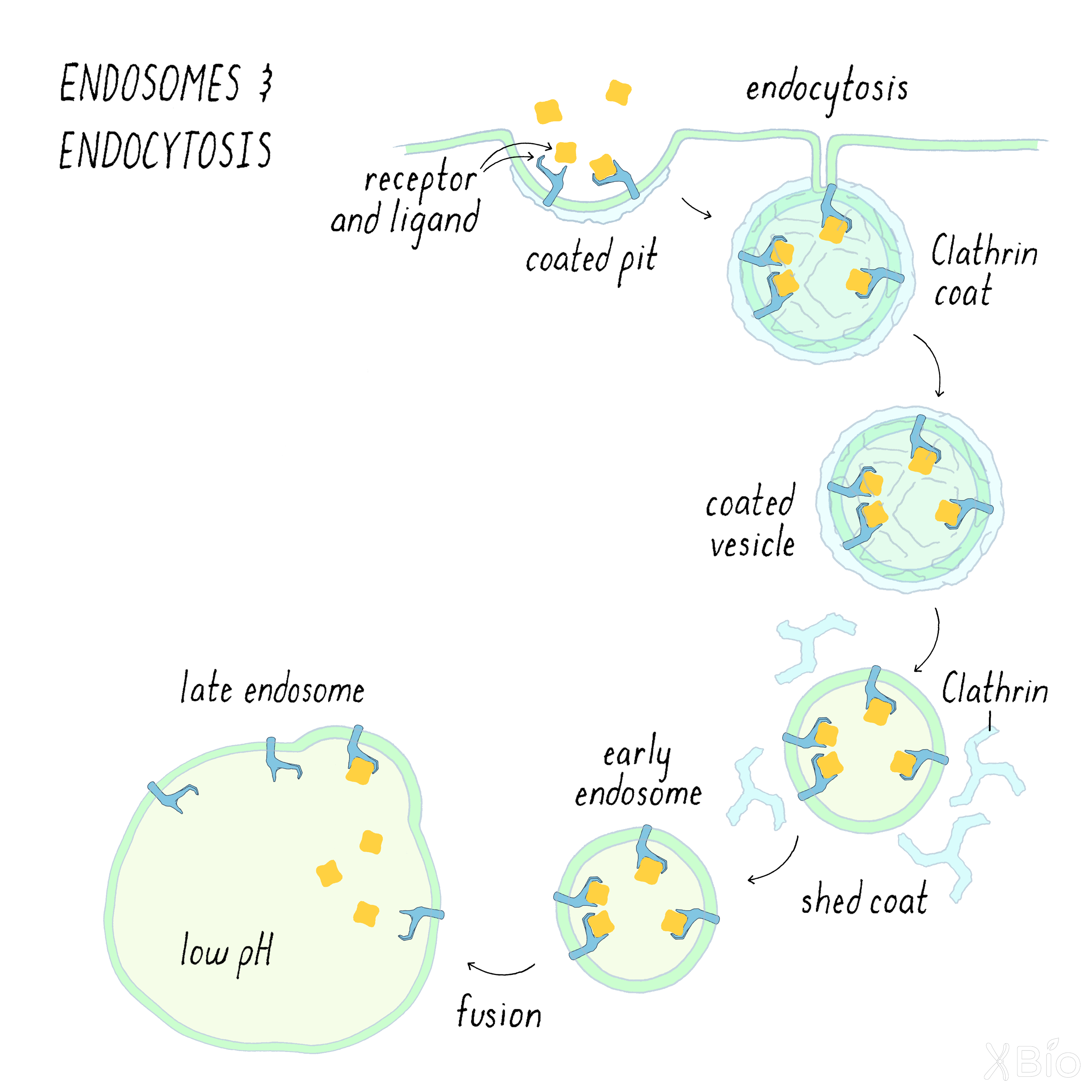
Endosomes
Endosomes (Figure 30) are internal membrane vesicles that bud off the plasma membrane by a process called endocytosis. Cargo (such a membrane receptors and other proteins) is delivered into endosomes by multiple routes, a major one involving a coat protein called clathrin. The clathrin forms a coated pit, which then buds off to create a coated vesicle. After internalization, the clathrin falls off of the coated vesicle to create an early endosome. Early endosomes then fuse with a larger tubular-vesicular membrane called the late endosome.
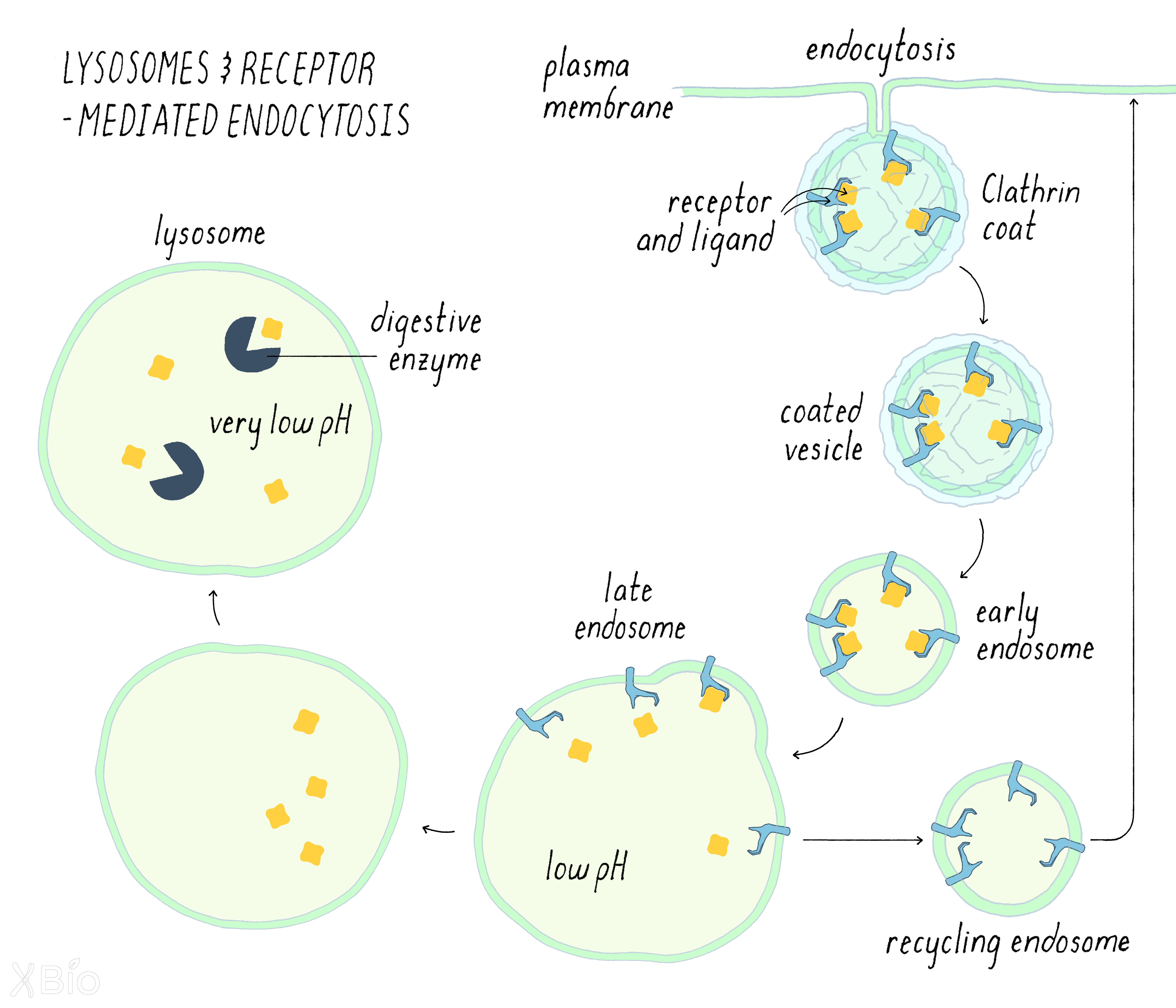
Lysosome
Lysosomes (Figure 31) are the major digestive organelle in cells. Lysosomes degrade substrates that originate from both outside and inside of cells. Extracellular substrates taken into the cell by endocytosis are delivered to lysosome via endosomes. Intracellular substrates, on the other hand, are delivered to lysosomes by a different pathway that involves autophagosomes (discussed below). To degrade materials, lysosomes contain numerous enzymes that breakdown various macromolecules, including proteases, lipases, and nucleases. Lysosomes are the lowest pH compartment in the cell, and many of these enzymes operate best at low pH.
More than 30 distinct lysosomal diseases in humans arise from defects in the function of one or more lysosomal hydrolases, attesting to the lysosome's essential role in clearance of materials from cells.
Now let us follow the full life cycle of one type of endocytosis called receptor-mediated endocytosis, which involves the lysosome. Ligand-bound receptors enter endosomes primarily through clathrin-mediated endocytosis. The ligand releases from its receptor due to the endosome's mildly acidic pH environment. The free ligand is then carried through the endosomal compartments. The late endosome then ultimately fuses with the lysosome, releasing its contents for degradation in the lysosome. Most of the receptors, on the other, return to the plasma membrane in a process called "recycling."
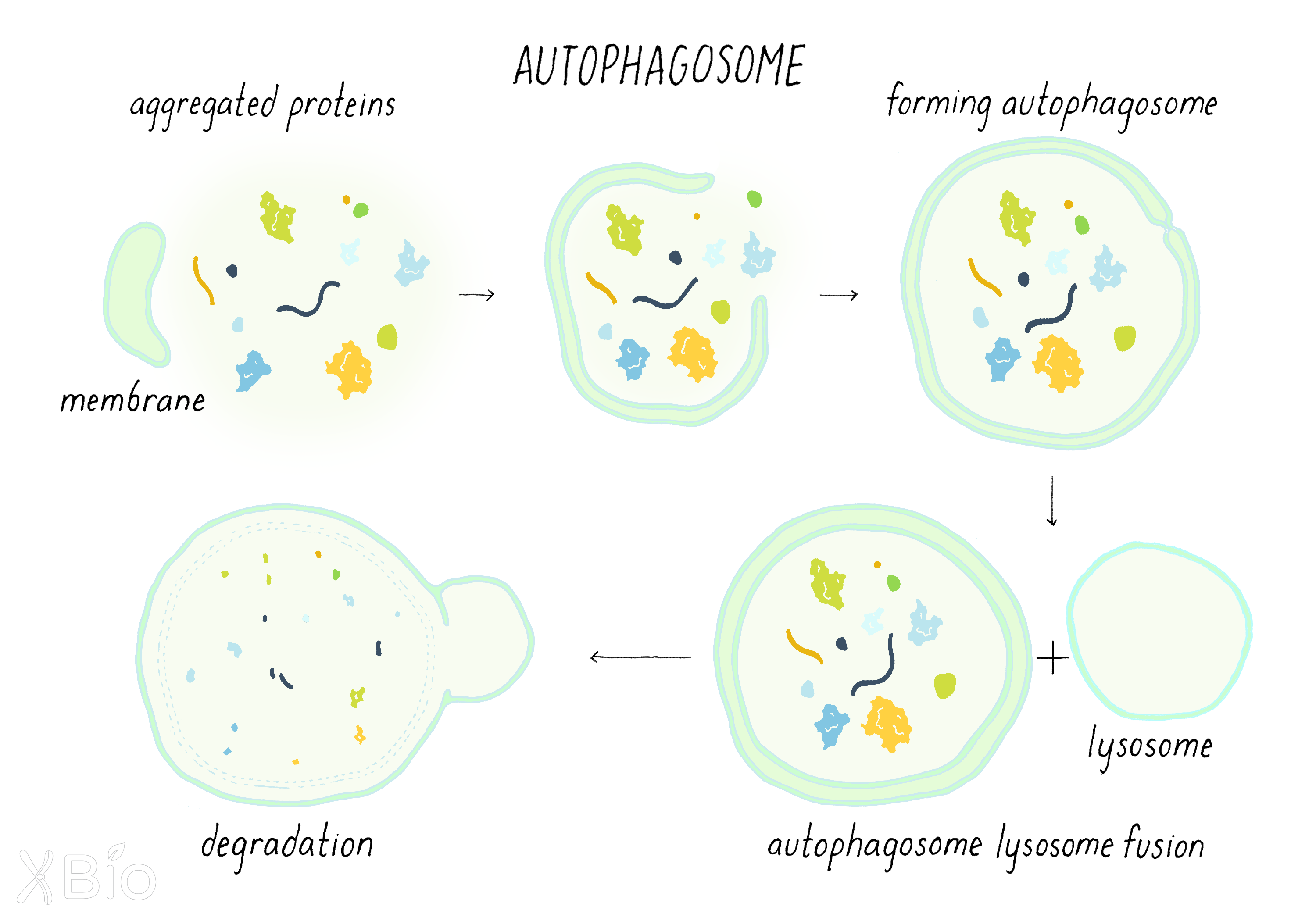
Autophagosomes
Autophagosomes (Figure 32) are double-membraned structures that engulf portions of the cytoplasm or other organelles and deliver this material to the lysosome for degradation. Autophagosomes form de novo by budding off from an organelle, usually the ER, mitochondria or endosome. A set of autophagy-related proteins drives this process by recruiting cytoplasmic autophagic components to target membranes. A double-membraned, cup-like structure then emerges from the target membrane to create the nascent autophagosome.
The autophagosome acts to envelope damaged proteins and/or organelles. It then delivers these damaged proteins/organelles for degradation by fusing with a lysosome. In addition to removing damaged components in the cytoplasm, autophagy is also a survival pathway for cells under starvation, as its degraded products can be recycled to serve as building blocks for new essential synthesis or energy for cell survival.
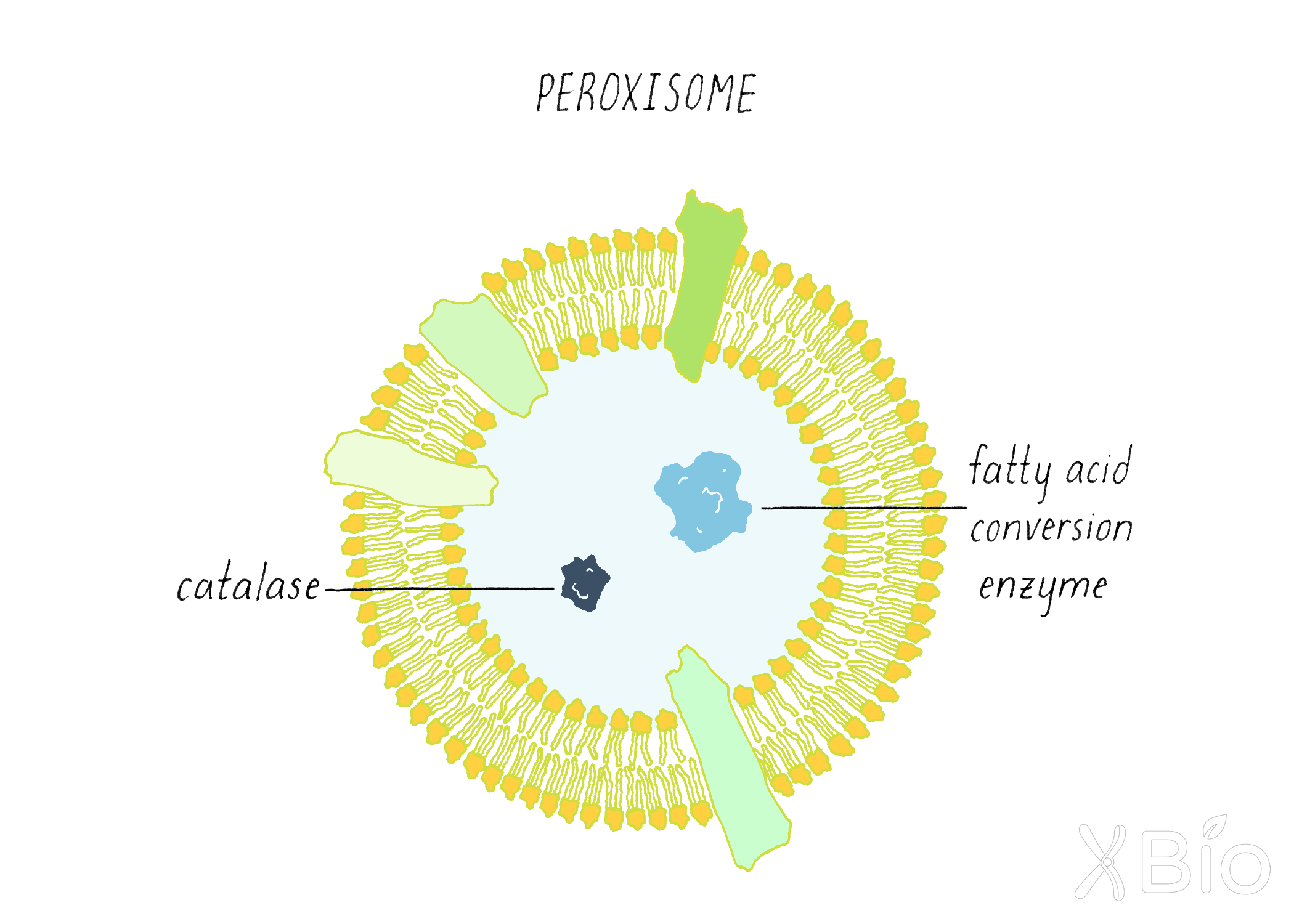
Peroxisomes
Peroxisomes (Figure 33) house critical enzymes involved in the conversion of very long chain fatty acids and branched fatty acids into simpler substrates for use by mitochondria in energy production. They also contain enzymes such as catalase that oxidize toxins and reduce reactive oxygen species (ROS). This function is especially important in liver cells, which are specialized for breaking down pollutants. The membrane of peroxisomes is derived from ER, but its enzymes are imported from the cytosol. Peroxisomes synthesize plasmalogens, a predominant phospholipid-type enriched in brain and required for forming myelin, the insulating layer around nerves that allows electrical impulses to transmit quicker.
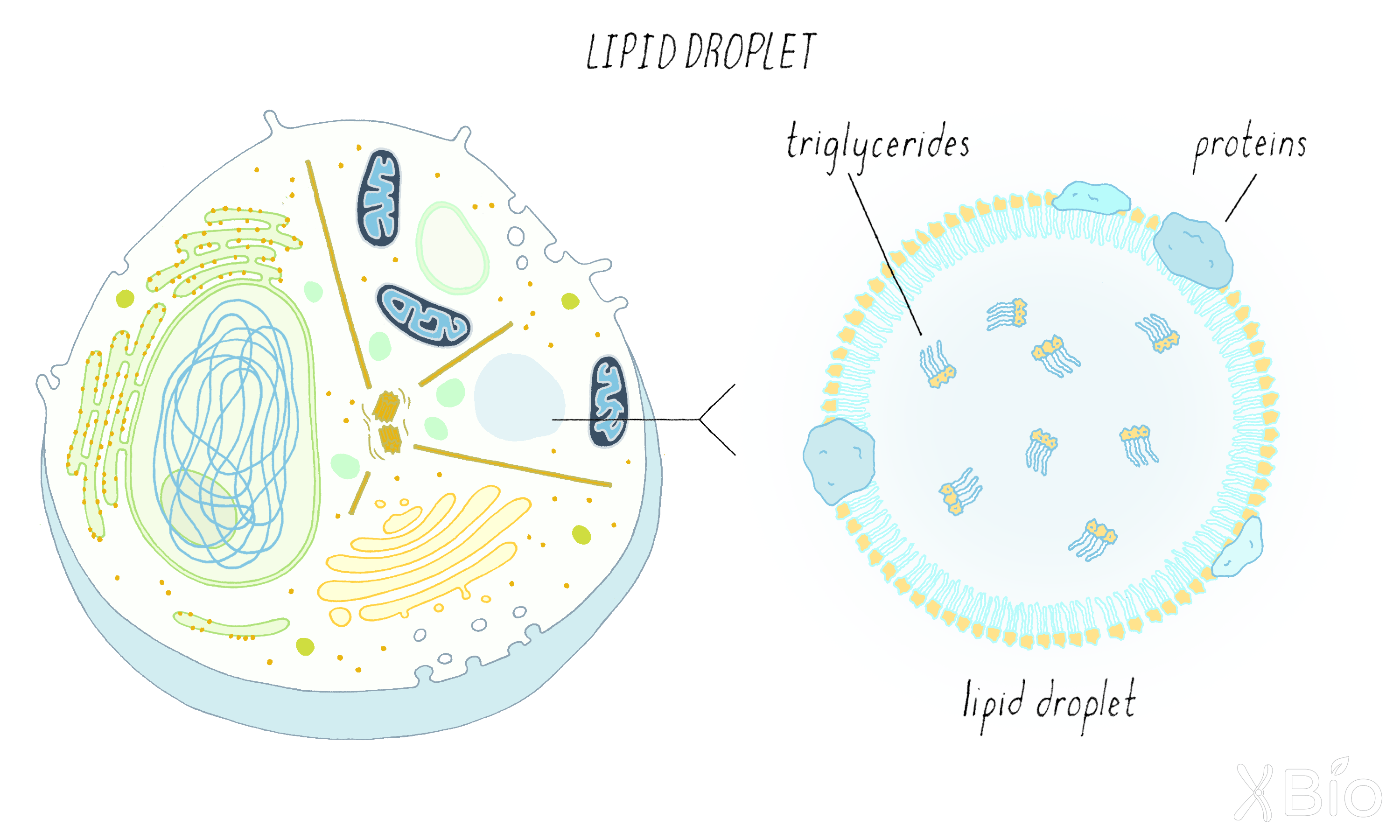
Lipid droplets
Lipid droplets (Figure 34) are the only organelle surrounded by a monolayer of lipids instead of a bilayer. The lipid droplet's lumen contains concentrated forms of lipids, particularly cholesterol esters (cholesterol bound to different fatty acids) and triglycerides (three fatty acids bound to glycerol). These stored lipids can be utilized either for membrane formation and maintenance, or as an energy source under starvation conditions. Lipid droplets derive from ER bilayers. Proteins can associate with the surface of the lipid droplet to regulate its activities, which include regulating lipid flux into mitochondria and peroxisomes.
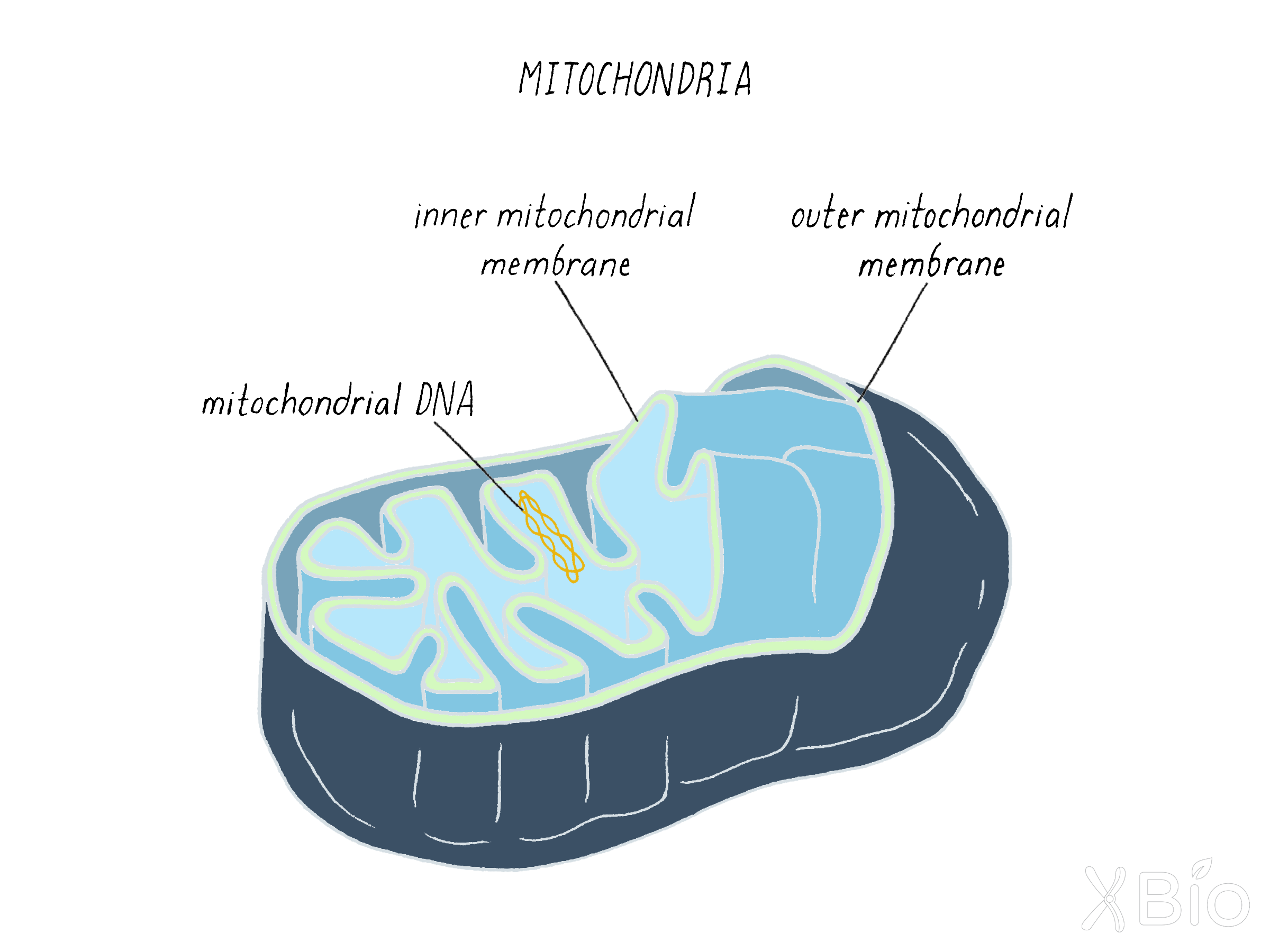
Mitochondria
Mitochondria (Figure 35) are the energy powerhouses of the cell. Unlike all other organelles (excepting ER), they cannot be formed de novo in cells. This fits with the idea that mitochondria were evolutionally derived from a bacterial endosymbiont engulfed by a host cell (discussed below).
Mitochondria are vital for numerous metabolic processes within cells. They have both an outer and inner membrane. The inner membrane surrounds the mitochondrial matrix, where most energy-related activities occur. These activities include: the Krebs cycle for consuming glycolytic products from the cytosol; beta oxidation for breaking down fatty acids into consumable elements; and oxidative phosphorylation for ATP production by the electron transport chain. These processes will be covered in another Narrative.
The matrix also houses the mitochondrial genome, which resembles bacterial genomes. The mitochondrial genome only encodes ~13 mitochondrial proteins, with the rest of its proteins encoded by the nuclear genome. These proteins use specialized import pathways to translocate across the two mitochondria membranes.
Although mitochondria are typically drawn as small peanut-shaped objects (e.g., Figure 35), they are often organized as tubules that are highly dynamic (Video 8). Mitochondria can frequently fuse and divide depending on the physiological state of the cell.
Exchange of materials between membranes
Organelles are not fully autonomous, and their activities must be integrated to benefit the whole cell. This is achieved through mechanisms that exchange materials between organelles.
As we have seen in the Journey to Discovery, one mechanism is the vesicular transport between organelles, which involves budding of vesicles from one organelle followed by fusion with another.
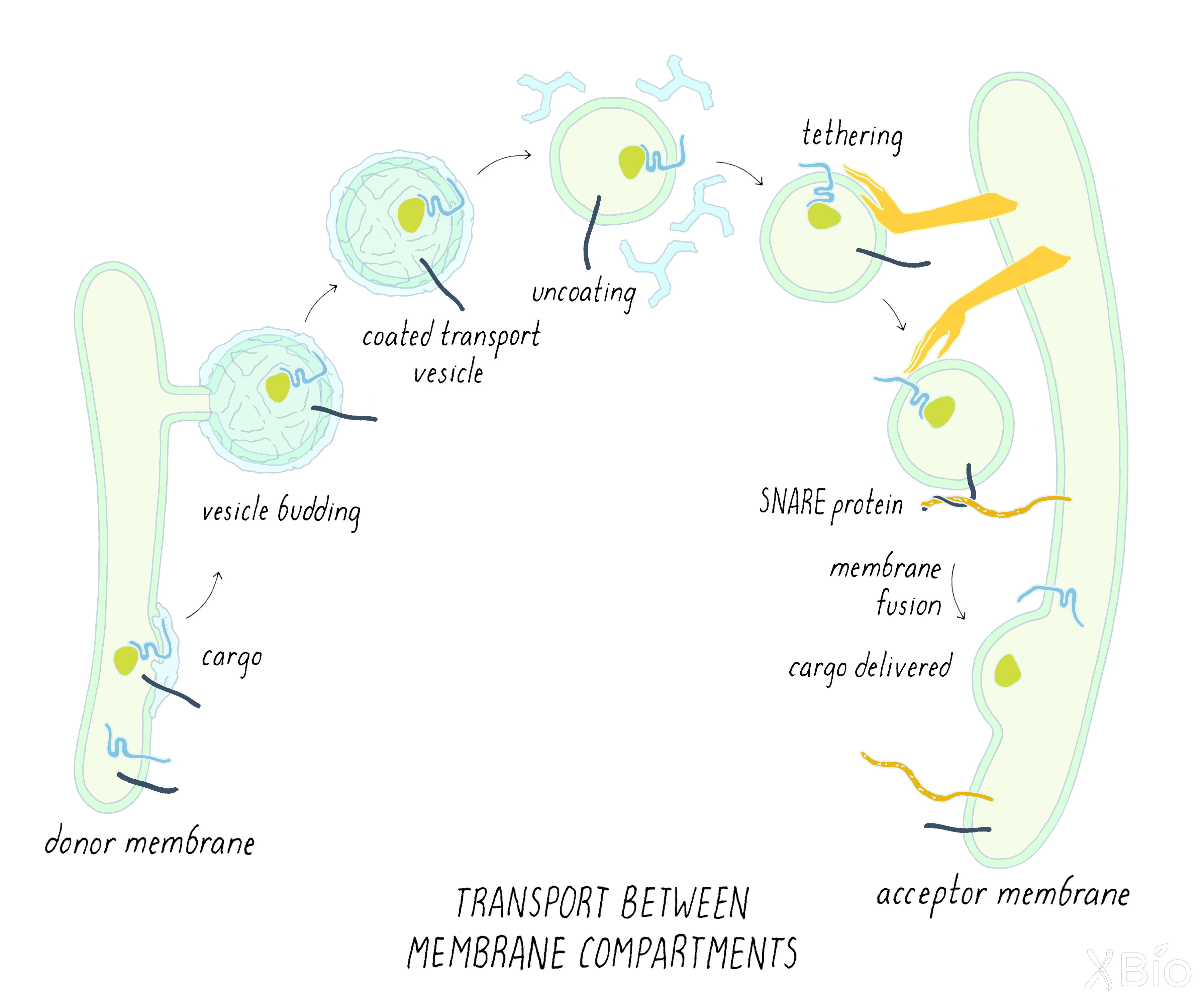
Membrane trafficking of cargo from one compartment to another
The discoveries of Schekman and Rothman led to the establishment of a general paradigm for the mechanisms of vesicular transport, applicable not only to the secretory pathway but also to other intracellular trafficking processes such as endocytosis. The steps in the process are indicated below (Figure 36):
1) A transport vesicle is first sculpted from the donor membrane by the assembly of a protein coat. The coat promotes vesicle budding while also selecting for specific cargos that will be incorporated into the vesicle.
2) After scission from the donor membrane, the vesicle loses its coat and moves through the cytoplasm toward an acceptor membrane.
3) Tethering factors associated with the acceptor membrane then capture the vesicle. Proteins called SNAREs then bring the very membranes very close together and trigger the fusion of the two membranes bilayers into one. This fusion process completes the delivery of cargo into the acceptor compartment. Specificity in this vesicular transport mechanism is determined by the use of different types of SNAREs, which mediate fusion with a particular target membrane.
Flow of cargo and lipid within this system (i.e., ER-to-Golgi-to plasma membrane and plasma membrane-to-endosome-to-lysosome) is balanced by recycling pathways (Figure 36). Without such retrieval pathways, organelles could not continue to maintain their unique lipid and protein composition.
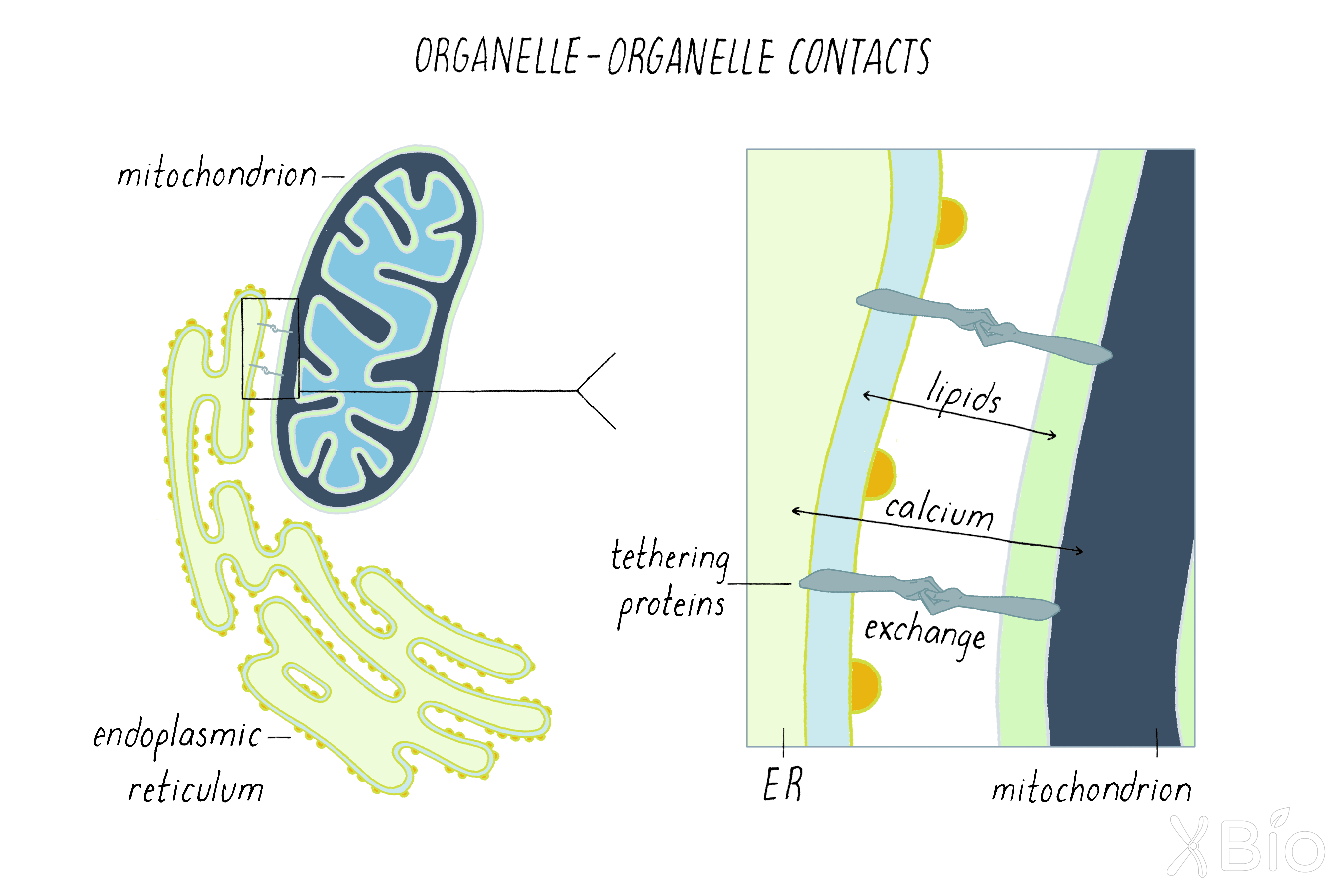
Direct contacts between organelles
In addition to exchanging materials through vesicular carriers, the large membrane organelles also can exchange metabolites and lipids through sites of very close association (called contact sites; Figure 37). This is mediated by tethering proteins that bring the membranes of two organelles in close proximity without fusion and permit other proteins to transfer metabolites such as calcium and lipids. This exchange plays critical roles in coordinating membrane growth and signaling activities of organelles within cells.
For example, mitochondria form contact sites with the ER to obtain both calcium and lipids under glucose starvation. Because contact sites are often difficult to see by conventional microscopy due to the crowded environment of the cytoplasm, they have only become more recently discovered through advanced imaging technologies and other methods. In fact, it has now been discovered that all cellular organelles make continuous contacts with the ER. Through such contacts, newly synthesized lipids in the ER are able to distribute to membranes of other organelles in the cell.
Defining the identity, shape, and location of a membrane compartment
Organelle identity
Secretory and endocytic pathway organelles exhibit an overall gradient in their sterol/sphingolipid compositions. This gradient in lipid composition gives these organelles distinct membrane fluidity characteristics that are important for their overall functions. The gradient is the lowest in the ER and increases sequentially from the Golgi to plasma membrane to endosome to lysosome. Low sterol/sphingolipid levels in the ER enables the ER to efficiently translocate proteins across its membranes. High sterol/sphingolipid concentrations in the plasma membrane allows it to be a better barrier to extracellular molecules. Even higher levels of sterol/sphingolipids in the lysosome makes its membranes stiffer and less impermeable, which is important for preventing the lysosome's hydrolytic enzymes from leaking into the cytoplasm and causing havoc. Intermediate concentrations of sterols/sphingolipids in the Golgi apparatus and endosomes aid in these compartments being membrane sorting stations.
The lipid gradient also helps direct newly synthesized transmembrane proteins to specific organelle destinations. This occurs because organelle membranes with low sterol/sphingolipid levels have thin bilayers and thereby attract proteins with short hydrophobic transmembrane domains. By contrast, areas with high sterol/sphingolipid levels have thicker bilayers and attract proteins with longer transmembrane domains.
Small GTPases, including Rabs, Arfs, and Sar1 proteins also play important roles in mediating trafficking and maintaining organelle identity. GTPases are recruited onto organelle membranes in their GTP-bound state (Figure 38). There they bind and affect the activity of other proteins, including coat proteins (COPI, COPII, and clathrin) that bud vesicles and proteins that bind and select cargos that go into vesicle carriers. These proteins also help to maintain organelle identity by recycling proteins and lipids recycled back to the original organelle after a deliver has been made. Cells typically use one Sar1 (operating on the ER), several Arfs (operating in Golgi), and ~70 different Rab proteins (operating on many different organelles) to specify the numerous transport steps comprising their secretory and endocytic pathways.
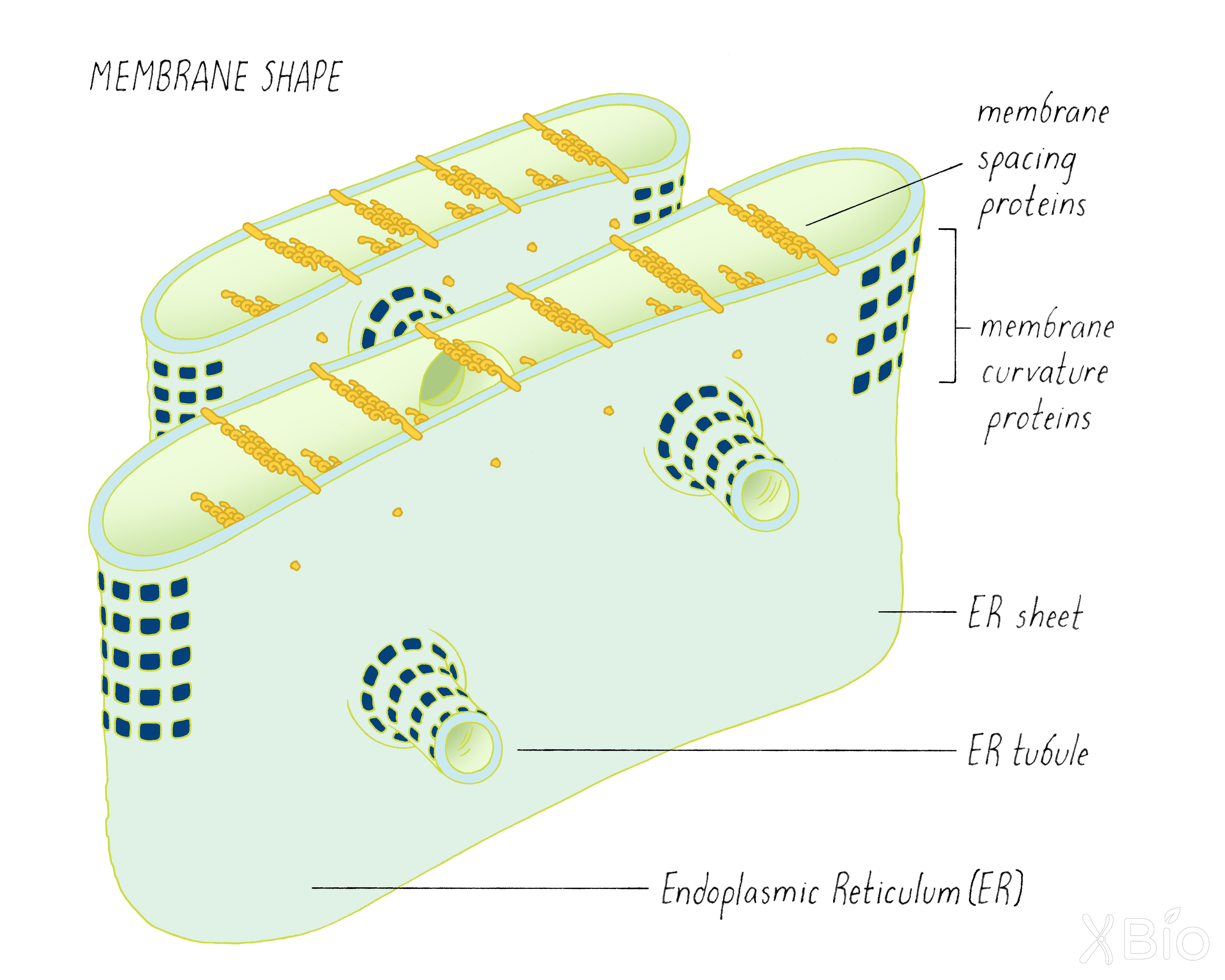
Organelle shape
Organelle shape is determined through the activities of embedded or peripherally associated membrane proteins (Figure 39). For example, the ER's tubular morphology is dependent on the presence of proteins that stabilize curvature. Proteins that bridge across a membranes, on the other hand, determine the spacing of ER cisternae. The Golgi's stack-like morphology, on the other hand, is thought to be maintained by other types of proteins that interact with each other across different cisterna.
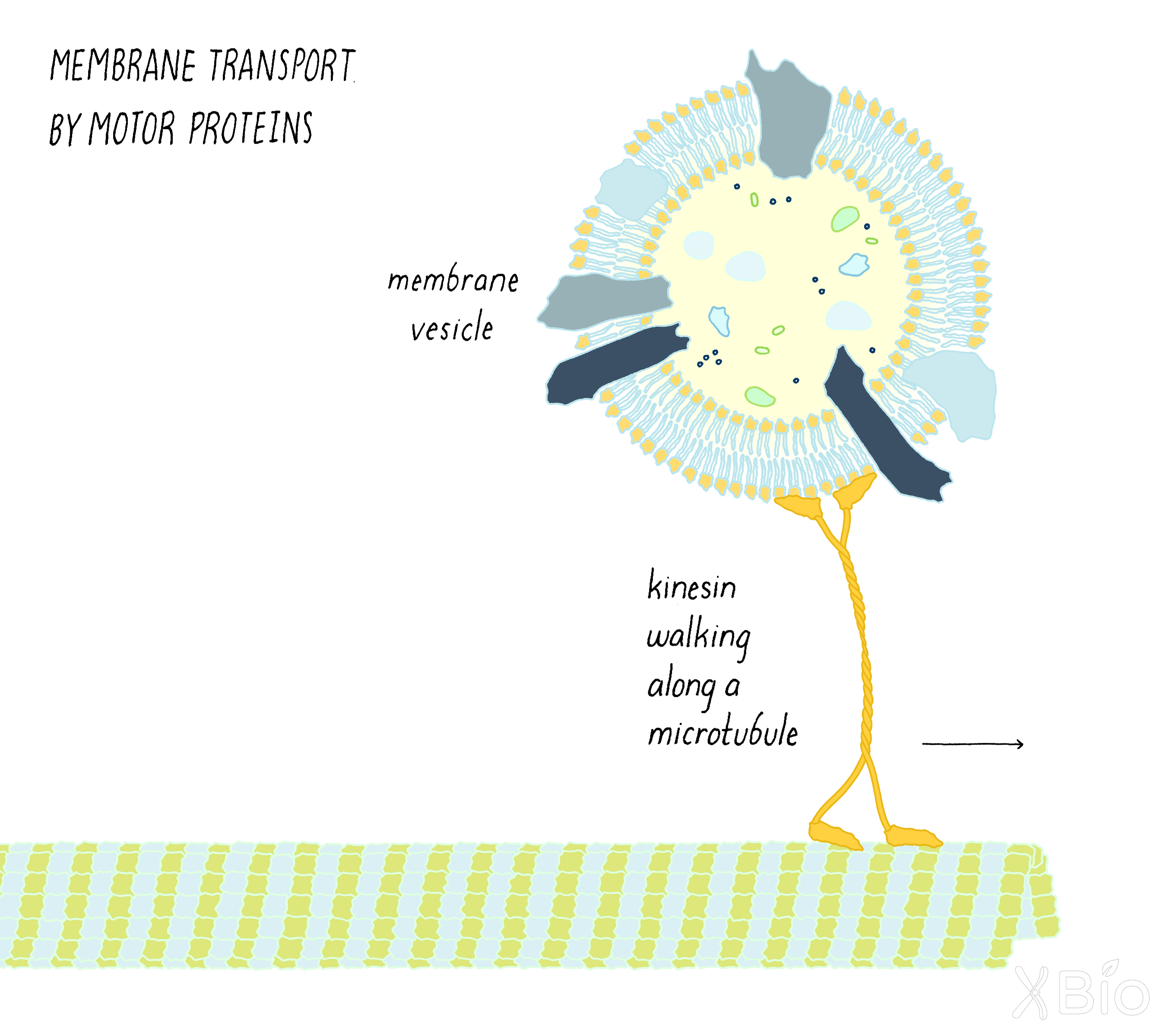
Organelle location and movement
Other proteins, called motor proteins, enable organelles to translocate to different locations within the cell (See the Key Experiment on motor proteins by Vale; Figure 40). Motor proteins walk along either a microtubule or actin cytoskeletal track. Highly motile lysosomes, for example, move in toward the nucleus using the dynein motor protein. They travel toward the periphery of the cell using kinesin motor protein. Transport vesicles travel between the ER and Golgi or Golgi and the plasma membrane through the directed transport by motor proteins.
Evolutionary origins of intracellular membrane systems
How did eukaryotic cells evolve internal membrane compartments? This is a challenging question because eukaryotic traits cannot be traced back in time beyond the last eukaryotic common ancestor (about two billion years ago), and this common ancestor already likely had all nine organelles seen in modern eukaryotes (i.e., ER, Golgi, mitochondria, etc.).
However, some theories suggest that eukaryotes originated when an ancient archaeal host cell engulfed and then sustained a bacterial endosymbiont, which went on to become mitochondria. This momentous event likely created the first primitive eukaryotic cell (Figure 41).
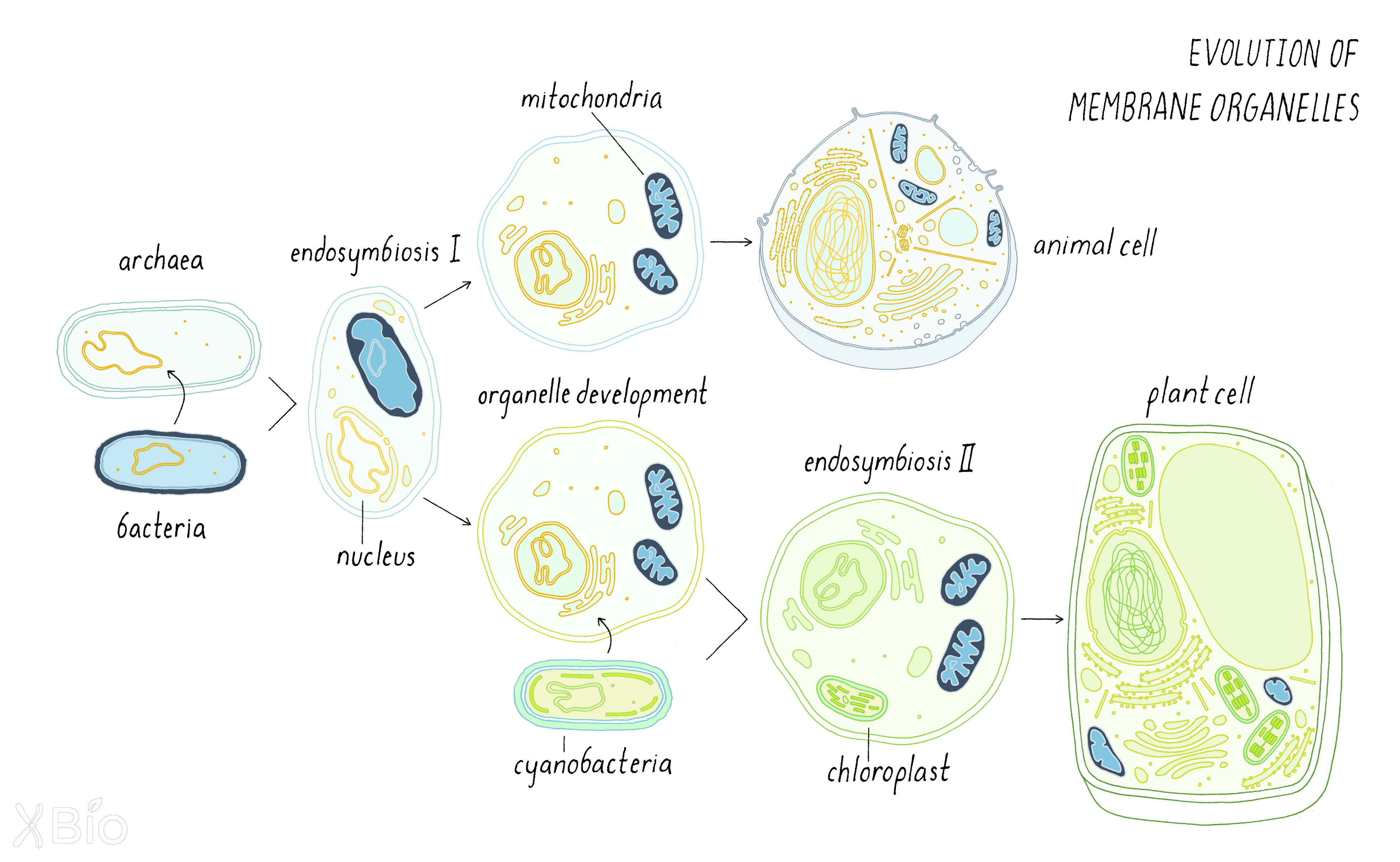
This merging of two prior independent life forms would have led to the chaotic mixing of molecules from the endosymbiont bacterium with the archaeal host with several profound consequences. First, lipids from bacteria (including sterol-like lipids called hopenoids and sphingolipids) would have mixed with the phospholipid-rich membranes of the archaeal host. These mosaic membranes may have produced different lipid subdomains and gradients in membranes. Proteins that recognize different lipid environments, GTPases, and other proteins, may have evolved to create membrane trafficking pathways.
One can only speculate on the order of events in the development of eukaryotic organelles. Initially, the first mosaic membranes may have been generated around the DNA to create the nuclear envelope. This may have served a beneficial role in keeping ribosomes away from DNA, acting as a barrier to separate transcription from translation. This feature may have afforded more layers of regulating protein expression. Once the nuclear envelope evolved, a simple extension of the nuclear envelope's surface would have produced the ER. Lipid mosaicism (different lipid domains within the membrane) in the ER, in turn, would have allowed it to produce transport intermediates that gave rise to the Golgi apparatus. Lateral segregation of proteins at the plasma membrane perhaps prompted endocytic pathways to evolve for taking up extracellular materials, another powerful innovation of eukaryotic cells.
We have learned a lot about the internal membranes of eukaryotic cells, but we have only scratched the surface. We still are learning more about what they do and how their unique identities are established and maintained. In the Frontier section, I will discuss a new EM microscopy technique that is providing an unprecedented 3D view of organelles inside of cells.
Part III: Frontiers —
A Return to Electron Microscopy for Visualizing Membrane Organelles in 3D
Electron microscopy (EM) is not new. We learned in the Journey to Discovery section how Palade, Porter, and Claude used EM to first visualize the many internal membranes of eukaryotic cells. It often said that the discoveries, which were made possible by EM, gave birth to the modern era of cell biology, similar to how the DNA double helix launched modern molecular biology. The awarding of the 1974 Nobel Prize in Physiology or Medicine for their "discoveries concerning the structural and functional organization of the cell" corroborates this statement. What then is left to improve with EM?
The EM images that Palade and colleagues were very thin 2D images, so just a tiny sliver of a very complicated 3D cell. As we learned in the Knowledge Overview, the shapes of membrane organelles have complex shapes and they contact one another and exchange materials. Understanding these aspects of membrane organelle function will require 3D imaging at high resolution. A new technological revolution in EM is turning this dream into reality.
Creating 3D images of the cell using electron microscopy
Electron microscopy allows for much higher resolution than light microscopy. However, electrons do not penetrate far into biological samples. Thus, a single EM snapshot is limited to a thin slice. In this case, there is only one way to create a 3D view of a cell—take many sequential 2D images through a biological sample and then assemble them computationally into a 3D rendering of a cell.
The notion of creating a 3D image through a series of slices is called tomography (tomo meaning slice in Greek). You or a friend may have had a medical CT (computed tomography) scan. This is a type of tomography for creating images of the human body, slice-by-slice. In this case, X-rays are used to create cross-sectional images at different angles, which then can be recombined computationally to recreate a 3-D image of a human organ or a knee. The development of CT scanning was discovery honored by the 1979 Nobel Prize in Physiology or Medicine.
Scientists also want 3D information of cells, but they are much smaller than organs. Electron microscopy is the answer. There are several ways of creating 3D views from electron microscopy slices. One method for achieving this is called focused ion beam (FIB) scanning electron microscopy (SEM). We are using FIB-SEM to create 3D images of cells at Janelia Research Campus (Video 9). Other groups at Janelia are using FIB-SEM to reconstruct images of the >100,000 neurons in a fly brain. We will discuss how this technique works next.
Focused Ion Beam Scanning Electron Microscopy (FIB-SEM)
Focused Ion Beam Scanning Electron Microscopy (FIB-SEM) offers high-resolution imaging in 3D. Let us discuss the various steps in making such a 3D image.
Preparing the sample
First, the sample must be "fixed," so that all cellular processes stop, but the intracellular structures are preserved. Fixing can be done with chemicals, which was the method used at the time of George Palade. However, now there are faster and better ways to preserve cell structure. A method of choice is high-pressure freezing.
As depicted in Figure 42, cells are cultured in a dish and then are suddenly exposed to high pressure (~2100 bar) and low temperature for a fraction of a second. For comparison, the pressure experienced by a scuba diver at a depth of 50 meters (150 ft) is only 6 bars. The high pressure reduces the freezing point, which prevents the formation of ice crystals that cause damage intracellular structures. The frozen cells are then taken through a process of freeze-substitution, which means removing all water (dehydration). The sample is slowly brought back up to room temperature in the presence of a heavy metal such as osmium, which fixes the structures in place. Osmium (an electron-dense metal) also binds to molecules, particularly to membranes, which provides better contrast for imaging with electrons from the microscope. After staining, the sample is then embedded in a plastic resin. The plastic embedded cells are removed from the dish and then are ready for the next step of FIB-SEM imaging.
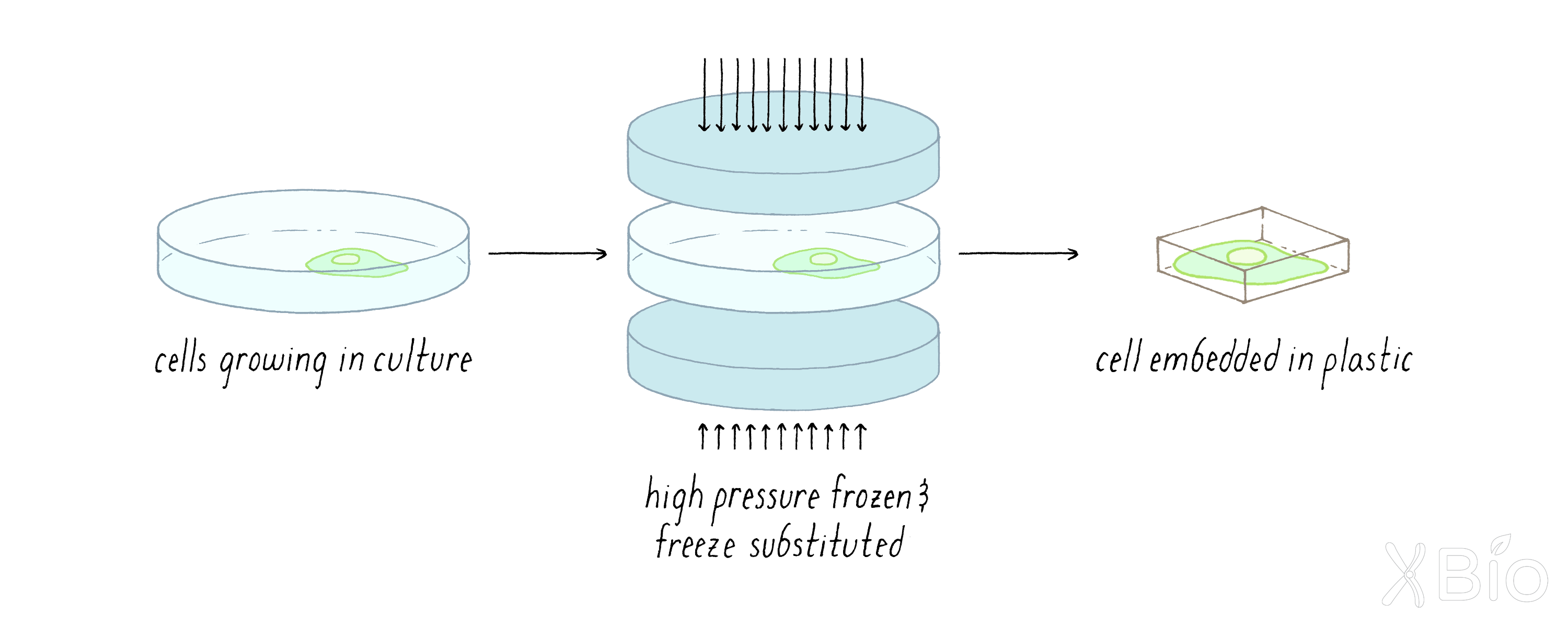
Milling and imaging
As the name suggests FIB-SEM is the combination of two techniques: 1) focused ion beam milling and 2) scanning electron microscopy imaging. Both tasks are performed by one instrument—the FIB-SEM microscope (Figure 43).

Milling is a common technique in machining in which a rotary cutter is used to remove material from a piece of metal or wood. Milling in FIB-SEM is similar, but it operates on a very different nanometer scale. In the milling step, a focused beam of very high energy ions (gallium) is focused on the cells embedded in the plastic. By scanning up and down, the high energy focused ion beam (FIB) precisely vaporizes a small layer of the material, typically between 2 and 8 nm.
In the imaging step, a focused electron beam is scanned across the block surface. The electrons are scattered off of the block surface in a way that can be imaged by the microscope.
To create a 3D image, the FIB-SEM microscope operates through alternating imaging and milling cycles (Figure 44). An image is acquired using SEM. Next, the FIB ablates a small layer. Another SEM image is acquired of the newly exposed surface. Then another layer ablated. The process repeats for many thousands of imaging and milling cycles for a whole cell.
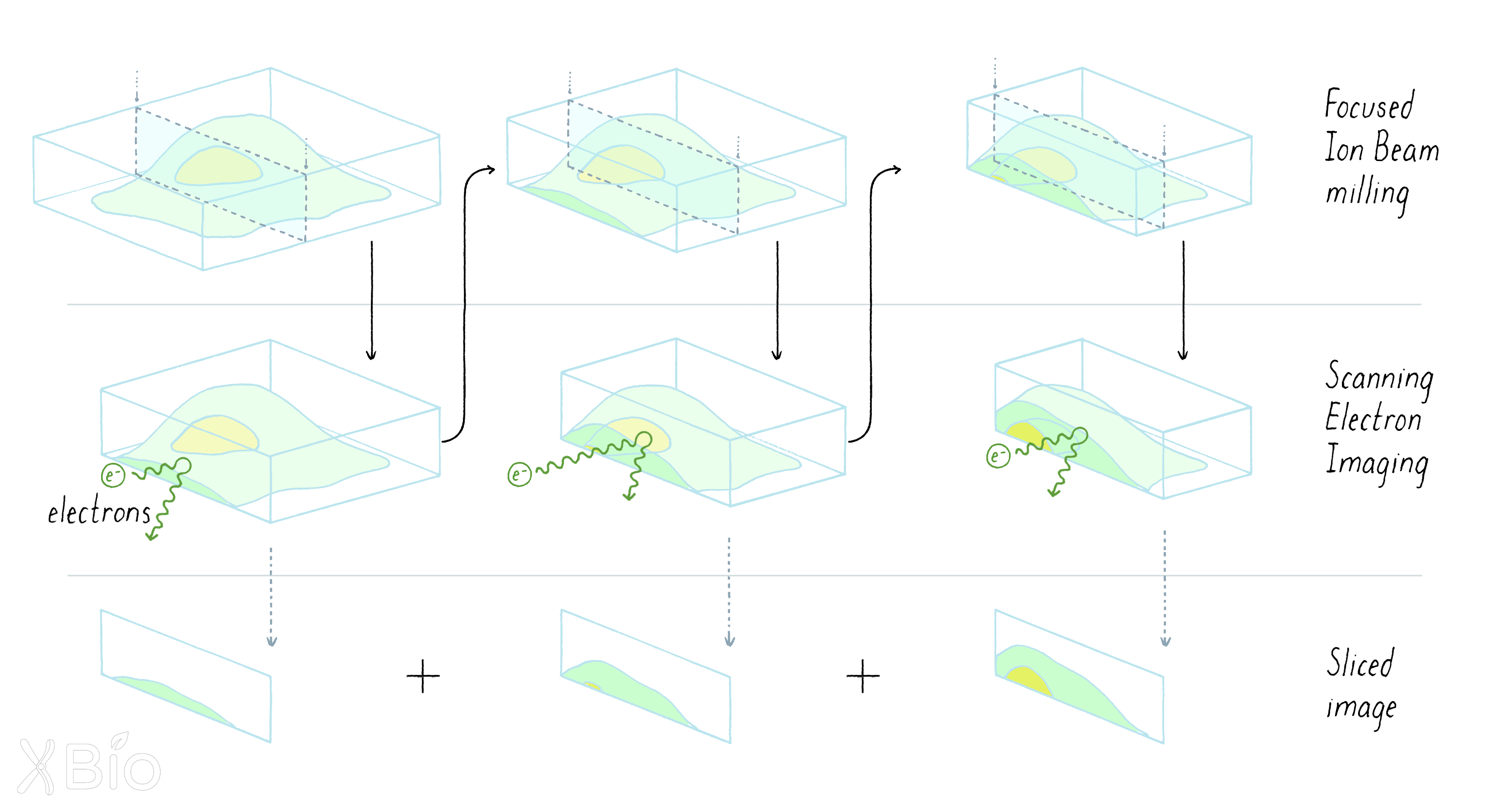
Next, all of the image slices have to be assembled into a 3D image. This is tricky to do properly. First, computational methods are used to fine tune the alignment between each slice. Then all of the slices are integrated into a 3D image data set. The amount of information that is stored within a single FIB-SEM data set is mind-boggling. At the end of Video 9, a 3D rending of a single cell (~8,000 cubic microns in size) is shown. This data set contains 122,265,600,000 voxels (a voxel is a volumetric pixel). That's 100 billion units of information that need to be analyzed. Thankfully, modern computers and computational scientists are up for this challenge.
The next step is to find interesting structures inside of the cell, such as membrane organelles. These structures have boundaries defined by the membrane bilayer that surrounds the organelle. Defining boundaries of objects inside of the cell, based on its staining intensity and morphology, is referred to as segmentation. This can be done manually, but it would take 50 years for one trained person to perform a complete segmentation of one single cell. Currently, scientists are using computers and machine learning to accelerate this process.
Finally, the segmented images have to be interpreted. What segmented structure is the Golgi? Are there new organelles that might be discovered? Can one see new substructures within or on the surface of organelles? The interpretation of these rich 3D images may require new computational strategies.
FIB-SEM is a good example of how biologists (who pose questions of how cells work), engineers (who build new milling microscopes), and computational scientists (who develop strategies for processing vast amounts of data) are working together to make new discoveries in biology.
A new era of discovery
New technologies allow scientists to learn new things about biology. We now have the capability to not just look at the ultrastructure of a couple of organelles but are poised to examine all of the organelles within a single cell at very high resolution. This work is still in progress, but promising results are already emerging (see Video 9).
The beauty of FIB-SEM is we are not limited to just a single cell. What if we thought bigger? What if we could image the entire brain of an organism? This effort is already underway. Researchers are imaging the entire brain of an adult Drosophila melanogaster and segmenting all of the cells within the data to yield a "wiring diagram" of the entire brain.
Imagine now, if we were able to combine information about the wiring of the adult fly brain with the ultrastructure of all organelles. We might be able to start connecting behavior of animals, to wiring diagrams of neurons, to organelle interactions and their distribution within each neuron.
Closing Thoughts
Numerous human diseases are associated with defects in the functioning of one or more of the cell's nine organelles. As noted in the introduction, viruses can hijack organelles and their vesicle trafficking pathways to enable viral uptake, multiplication, and release from cells. Other diseases may arise from organelle dysfunction. Bone disorders have been linked to defects in ER morphology and ER to Golgi trafficking. Parkinson's disease, diabetes, and deafness are all thought to arise from defects in mitochondria function. While we have some ideas for how disrupted organelle function might contribute to such large-scale disease states, we are still largely in the dark.
Part of the problem is that while we know the list of organelles and their functions, we still do not know how organelles vary in their function and distributions across different cell types. Only recently, for example, was it realized that organelles in brain neurons and their neighboring astrocytes (a type of neuron-associated glial cell) respond differently to excitotoxicity (too much electrical activity). Lipids in both cell types are damaged by the excitotoxicity; however, astrocytes protect themselves by transferring these lipids to lipid droplets for storage and ultimate mitochondrial metabolic consumption, whereas neurons are unable to do this. They instead secrete the toxic lipids in particles that nearby astrocytes engulf and then consume. Anything that interferes with this exchange process, as may occur in some neurodegenerative disease, leads to neuronal cell death.
Could one develop new strategies that minimize damage to membrane organelles during disease or enhance their functions to potentially prevent or delay disease? Such a future might be possible, but we need to know more about what organelles do. The usual descriptions of membrane organelles in biology textbooks make it seem that we understand everything about them. However, in my view, a large number of questions remain unanswered. For example, most of the work on organelles has focused on simple organisms such as yeast (e.g., Randy Schekman's work in the Journey to Discovery) or in mammalian cancer cells growing in tissue culture (e.g., Jim Rothman's work in the Journey to Discovery). However, we still lack a comprehensive view of organelle distribution and activity for the numerous different cell types that make up the human body. We also do not understand the many rules governing the "life cycle" of an organelle, that is, how they grow, are maintained, and then turn over. There are lots of questions that fascinate me and call out for more experiments.
So, how do we approach these new questions? My view is that we need new technologies that can peer into the dynamic activities of intracellular organelles. For example, what if we could build sensors that report on mitochondrial energy states, inter-organelle trafficking pathways, Ca2+ handling, membrane lipid toxicity, and membrane voltage in living cells? What if we could read-out this information in a mouse with neurodegenerative disease or diabetes? My bet is that we would understand much more about what membrane organelles are doing and how organelle dysfunction gives rise to human diseases.
These capabilities might seem impossible today. However, the history of cell biology shows that the impossible eventually becomes possible. Camillo Golgi did not have an electron microscope to study the Golgi apparatus, as did George Palade. Many of the biochemical tools that Jim Rothman used for his experiments on the Golgi were not available at the time of Palade. I took advantage of the green fluorescent protein to study the Golgi (see Key Experiment by Chalfie), a tool that emerged only after Rothman's original studies. This leap-frogging to new methods and tools continues. I now have the privilege to work with engineers who currently are making new microscopes and chemists who make new fluorescent probes. Through such efforts, I am confident that scientists will learn more about membrane organelles, and thereby provide new insights into tackling some of the most difficult diseases afflicting our species.
Acknowledgments
I thank Aubrey Weigel for her help in writing the Frontier section.
Dig Deeper
Dig Deeper 1: Temperature-sensitive mutations
Temperature-sensitive (ts) mutations are very useful for studying a biological process, since the effect of the mutation can be turned on or off by shifting the temperature. At the "permissive temperature," the function of the gene is normal and the organism behaves similar to the wild type organism without the mutation in the gene. However, at the "restrictive temperature," the mutation now exhibits a phenotype (a change in some trait). The phenotype is reversed when returned to the "permissive temperature." The restrictive temperature is usually a temperature that is higher (heat-sensitive) or lower (cold-sensitive) than the normal temperature at which the organism lives. Temperature-sensitive (ts) mutations can be produced not only in many microbes (bacteria, yeast) but also in more complex organisms such as flies.
What is the basis of this temperature-sensitivity? A mutation in a gene (which can occur naturally or be induced by chemicals or UV light; see Narrative on Mutations by Koshland) can produce a change in the amino acid sequence of the encoded protein. Frequently, a ts mutation changes just a single amino acid and usually one that is not absolutely critical for its function. However, that single amino acid change can affect the shape of the protein in a way that is dependent upon the temperature. The shape change might be subtle, but it can be enough to partially or completely inactivate the protein.
Temperature-sensitive mutations are rare. Most single amino acid changes do not affect the function of a protein at all. Of the ones that produce an effect, the vast majority will affect protein function (inactivating or sometimes activating) but not in a temperature dependent way.
A ts mutation can be particularly valuable in studying the function of a gene that is essential for the viability of an organism. The organism can be grown at the permissive temperature (so that the strain can be propagated). Then, the organism is shifted to the restrictive temperature to shut-off the function of the gene and examine what happens. Schekman used this approach to study the role of the sec-1 gene in the Journey to Discovery.
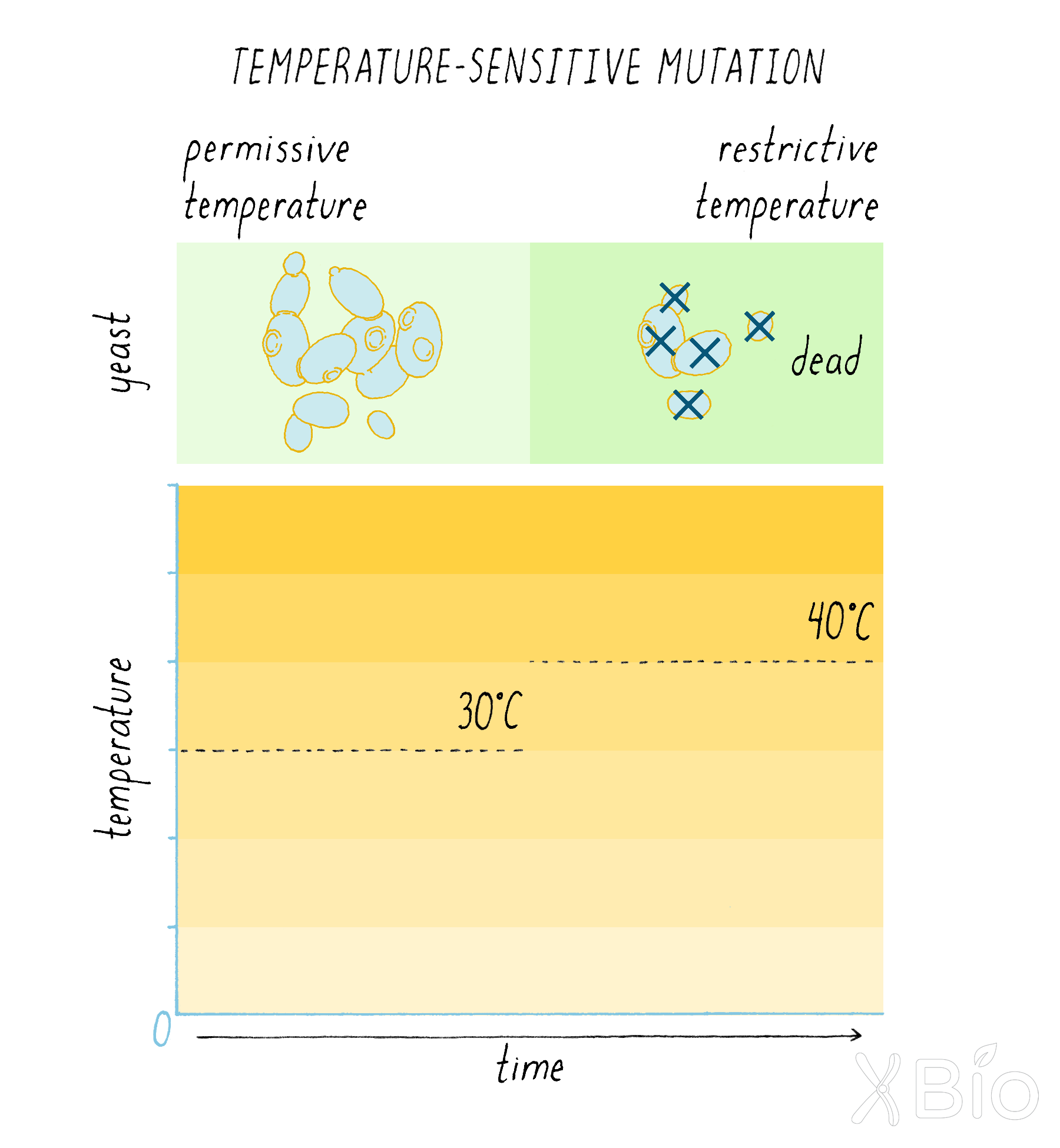
Dig Deeper 2: Immunoprecipitation
Immunoprecipitation, or also called immuno-pulldown, is a method for using antibodies (immuno) to separate, through centrifugation, particular proteins or sometimes organelles from the cell. Antibodies are special proteins made by the mammalian immune system that recognize a foreign antigen, such as a protein from an invading organism. A specific antibody will recognize a very specific protein. Traditionally, scientist made antibodies by injecting a protein into a rabbit, having the rabbit mount an immune response, then collect blood from the rabbit, and purify the antibody made against that protein. Now, there are also ways to make recombinant antibodies in bacteria or yeast.
The steps in immunoprecipitation or immuno-pulldown are depicted in Figure DD2, although there are alternative methods as well. First the cells are broken, releasing the interior content (a cell lysate). An antibody to a specific protein is attached to micrometer-sized bead and the antibody-attached beads are incubated in the cell lysate. During the incubation, a specific protein in the lysate will attach to the antibody on the bead. Next, the tube is placed in a centrifuge that spins the sample at high speed. The beads are large and heavy and, due to the centrifugal force, will move to the bottom of the tube much faster than the small proteins. After centrifugation, the top material (called the supernatant) is removed, which contains most proteins in the cell that did not bind to the bead. Then the material at the bottom (the pellet) is collected, which contains the antibody bound to the beads and any protein bound to the antibody (and also potentially any other proteins bound to the first protein).
The proteins bound to the beads can then be analyzed by a technique called SDS-PAGE—Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis. In this method, protein–protein interactions are broken apart, and the individual proteins are separated according to their size as the move through an electrophoretic field. The separated proteins can be visualized by stains or other methods. Although not precisely the same, the concept of running a gel is shown in this video for Agarose Gel Electrophoresis.
Immunoprecipitation was used in the experiment by James Rothman in the Journey to Discovery. In this experiment, he wanted to know how much of a radioactive sugar was attached to the G protein. To determine this, Rothman immunoprecipitated the G protein with a specific antibody and then could measure how much radioactive protein was specifically incorporated into the G protein.
Immunoprecipitation is widely used in biochemistry. In the addition to the type of experiment described above, it can be used to study interactions between proteins. By pulling down one protein, one can see what other proteins are bound to it and thus might be involved in a similar cellular function.
Explorer’s Question: In the immunoprecipitation experiment described above, how can one be sure that the additional proteins that come down with the beads are interesting protein interactions and not some kind of artefact? What controls would you do?
Answer: One control is just put in beads with a non-specific antibody—i.e., some antibody to another protein that is not even found in that cell (e.g., an antibody to a bacterial protein that is not found in a mammalian cells). You want to make sure that the “interesting” protein that you found is not just sticking nonspecifically to beads or antibodies in general. You also have to make sure that the antibody to your first protein is also specific. Antibodies sometimes can cross-react with multiple proteins. Then the extra proteins that you see might just be antibody cross-reactions with multiple proteins and not reflect “interesting” proteins interacting with your protein of interest. There are several methods, such as immunoblot analysis, that can help to determine the specificity of your antibody.
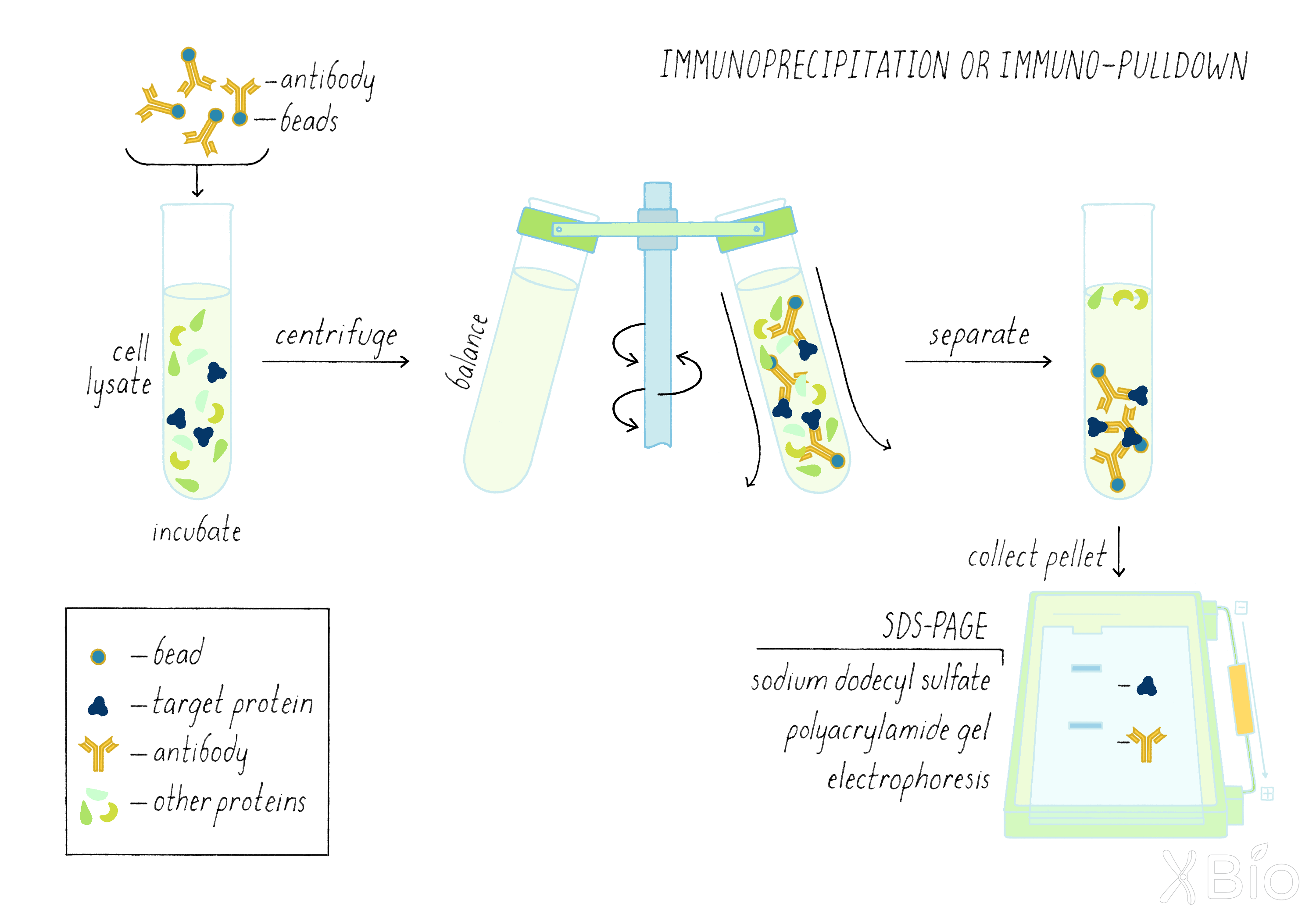
Dig Deeper 3: Exploring membrane dynamics using fluorescence recovery after photobleaching (FRAP)
FRAP is a powerful tool for investigating protein dynamics within organelles. It reveals the extent to which a fluorescently tagged protein can freely move (i.e., diffuse) or is immobile within a membrane. To perform FRAP, a region of a cell expressing a fluorescently tagged protein is photobleached with a high intensity laser. This process inactivates the fluorescence in the zone of the intense laser light, making those proteins no longer visible. If the labeled proteins are highly mobile, then molecules that have not been photobleached in the area surrounding the bleached zone will quickly move into and exchange positions with the photobleached molecules within this zone. This mobility is due the process of "diffusion" which causes molecules to move randomly about by thermal agitation. The process of diffusion leads to fluorescence recovery after photobleaching, in which the photobleached/darkened zone regains fluorescence over time. From the speed of fluorescence recovery, it is possible to calculate the molecule's diffusion coefficient. If recovery is slow or does not occur, this result would suggest that the molecules in the membranes of the organelle are restrained in some fashion (i.e., by being bound to a scaffold or spatially restricted in some fashion).
References and Resources
Guided Paper
Novick, P. and Schekman, R. Secretion and cell-surface growth are blocked in a temperature-sensitive mutant of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1979;76:1858–1862.
A classic paper on yeast genetics describes finding genes involved in protein secretion for the first time. This research led to Schekman receiving his Nobel prize in 2013.
DownloadReferences
- Jamieson, J.D. and Palade, G.E. Intracellular transport of secretory proteins in the pancreatic exocrine cell. II. Transport to vacuoles and zymogen granules. J Cell Biol. 1967;34:597–615.
- Balch, W.E., Murphy, W.G., Braell, W.A. and Rothman, J.E. Reconstitution of the transport of protein between successive compartment of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 1984;39:405–416.
- Matlin, K.S. and Caplan, M.J. The secretory pathway at 50: a golden anniversary for some momentous grains of silver. Mol Biol Cell. 2017;28:229–232.
- Pfeffer, S. A prize for membrane magic. Cell. 2013;155:1203–1206.
Resources
- National Human Genome Research Institute's Organelle Glossary. Available at: https://www.genome.gov/genetics-glossary/Organelle
This website gives a narrated overview of what organelles are as well as a diagram of some of the more common organelles in the cell.
- Jennifer Lippincott-Schwartz iBiology talk on "How lipids and cholesterol regulate trafficking across the secretory pathway." Available at: https://www.ibiology.org/cell-biology/lipids-and-cholesterol/
- Randy Schekman's iBiology talk on "Protein secretion and vesicle trafficking." Available at: https://www.ibiology.org/cell-biology/protein-secretion/
- Frontiers in Cell and Developmental Biology: The different facets of organelle interplay: An overview of organelle interactions. Available at: https://www.frontiersin.org/articles/10.3389/fcell.2015.00056/full
This review article published in 2015 summarizes the available data on the existence and composition of organelle interaction and the contact sites through which organelles interact.
- Nucleus Medical Media- 360° Guided Tour of the Cell (demo). Available at: https://www.youtube.com/watch?v=rKS-vvhMV6E
This animated video provides a brief tour of cell organelles and how they interact when a cell is functioning properly.
- Johnkyrk.com- Interactive Website on Cell Membranes. Available at: http://www.johnkyrk.com/cellmembrane.html
This website gives an overview of the structure, composition and function of cell membranes with some interactive components.
- Nature.com scitable: Cell Membranes. Available at: https://www.nature.com/scitable/topicpage/cell-membranes-14052567/
This website created by Nature Education gives an overview of the structure, composition and function of cell membranes.
- Nucleus Medical Media—Biology: Cell Structure, Available at: https://www.youtube.com/watch?v=URUJD5NEXC8
This video provides detailed animated visuals representations of what organelles within the cell look like as well as a narration regarding their structure and function.
- HarvardX- Mitochondria: the cell's powerhouse. Available at: https://www.youtube.com/watch?v=vkYEYjintqU&list=PLltdM60MtzxPl5IgreGlrUAzHQ8O0OoK3
This video provides a high quality animation and narration regarding the structure and function of the Mitochondria.
- HarvardX- Overview of cell structure. Available at: https://www.youtube.com/watch?v=XOaiWl-nW1k&list=PLltdM60MtzxPl5IgreGlrUAzHQ8O0OoK3&index=5
This video provides a high-quality animation and narration regarding cell structure including a brief overview of cell membranes and membrane proteins, ribosome-mitochondria interactions and vesicle packing and transport between the ER and Golgi as well as through the cytoplasm.
- HHMI Biointeractive- Pulse-Chase Primer. Available at: https://www.biointeractive.org/sites/default/files/HHMI%2520Pulse-Chase%2520Primer%2520-%2520Student.pdf
This document contains an overview of what a pulse-chase experiment is along with questions and activities regarding pulse-chase experiments and the concepts behind them.
- Jennifer Lippincott-Schwartz iBiology talk on "Membrane dynamics: photobleaching and photoactivation." Available at: https://www.ibiology.org/talks/photobleaching-and-photoactivation/